原始数据
wget https://raw.githubusercontent.com/pzweuj/practice/master/R/Hist_PCA_Deno/Allen_BrainAtlas_12regions_Microarray.txt
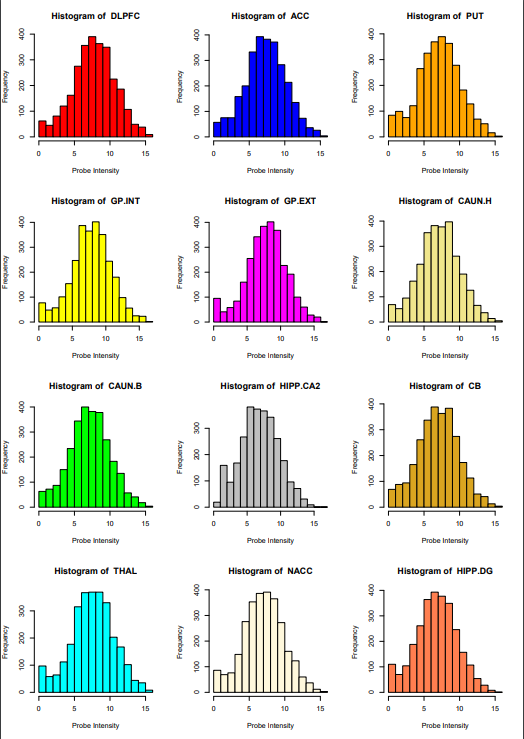
柱状图
这里一共是有12个区域,画12个图并且放到一张图上。
brain12 <- read.table("Allen_BrainAtlas_12regions_Microarray.txt", header=TRUE)
# 设置12种颜色
# 应该可以利用随机函数还有colors()函数来随机生成或者直接使用前12种颜色,更方便
col <- c("red",
"blue",
"orange",
"yellow",
"magenta",
"khaki",
"green",
"gray",
"goldenrod",
"cyan",
"cornsilk",
"coral")
# 设定输出目标
pdf("Histograms.pdf", width=8.27, height=11.69)
# 合并下面的图
par(mfrow=c(4, 3))
# 循环出图
for(i in c(1:12)){
xname = colnames(brain12[i])
hist(brain12[, xname], main=paste("Histogram of ", xname),
col=col[i],
xlab="Probe Intensity", ylab="Frequency")
rm(xname)
}
# 输出结束
dev.off()
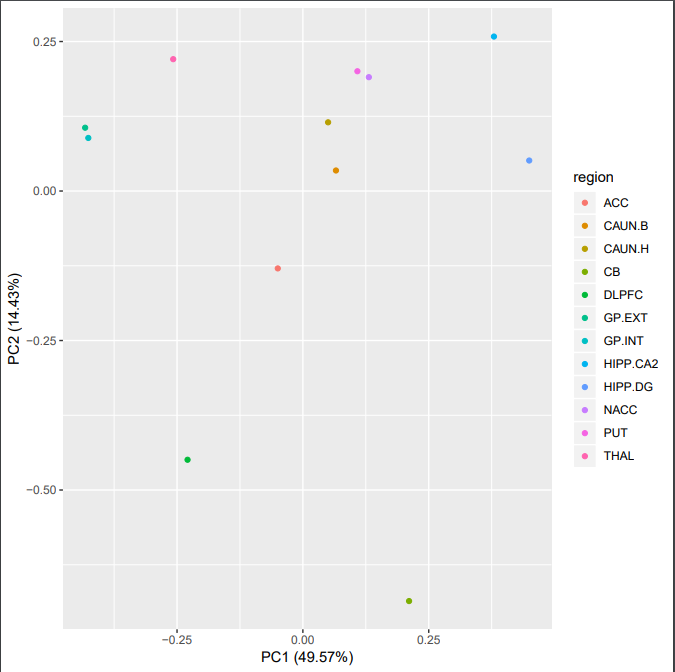
PCA图
使用ggfortify这个包来画。
# install.packages("ggfortify")
library(ggfortify)
# 前处理
brain12 <- t(brain12)
brain12 <- as.data.frame(brain12)
brain12$region <- rownames(brain12)
# 计算pca
brain12.data <- brain12[c(1:2817)]
pca <- prcomp(brain12.data)
summary <- summary(pca)
# 输出pdf
pdf("PCA.pdf")
autoplot(pca, data=brain12, colour="region")
dev.off()
# 输出summary
write.table(summary$importance,
file="PCA_summary.txt",
sep='\t', col.names=NA, quote=FALSE)
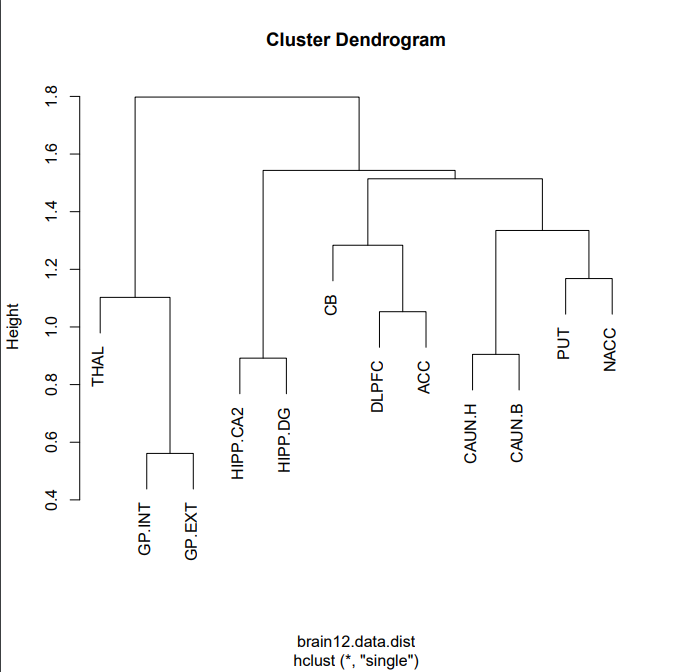
系统发育树
直接用内置函数了。
# 数据标准化
brain12.data.scale <- scale(brain12.data)
# 使用皮尔森校正
brain12.data.cor <- cor(t(brain12.data.scale), method="pearson")
# 计算距离
brain12.data.dist <- dist(brain12.data.cor)
# 创建系统发育树的类
hc <- hclust(brain12.data.dist, method="single")
# 画图
pdf("Dendrogram.pdf")
plot(hc, main="Cluster Dendrogram")
dev.off()