panel检测和外显子、全基因检测相比,有耗时短,价格便宜,针对性高等优点。 但是,缺点也是很明显的,比如说碱基个数不等的拷贝数/增添/缺失等突变,panel比较难测出来。 还有就是panel没有覆盖到就会有阴性的结果出现。还有就是panel都是基于前人的研究的,如果研究不准确,panel就会错。
而全基因,外显子等还有校正的余地。
这里以癫痫为例子,简单的介绍一个做panel的方法。 首先,我们要用到的数据库是OMIM,人类孟德尔遗传数据库。搜索癫痫,得到结果。 当然,单纯的epilepsy是不足以覆盖到所有癫痫基因的,我建议更准确的方法是找到所有癫痫的相关疾病。
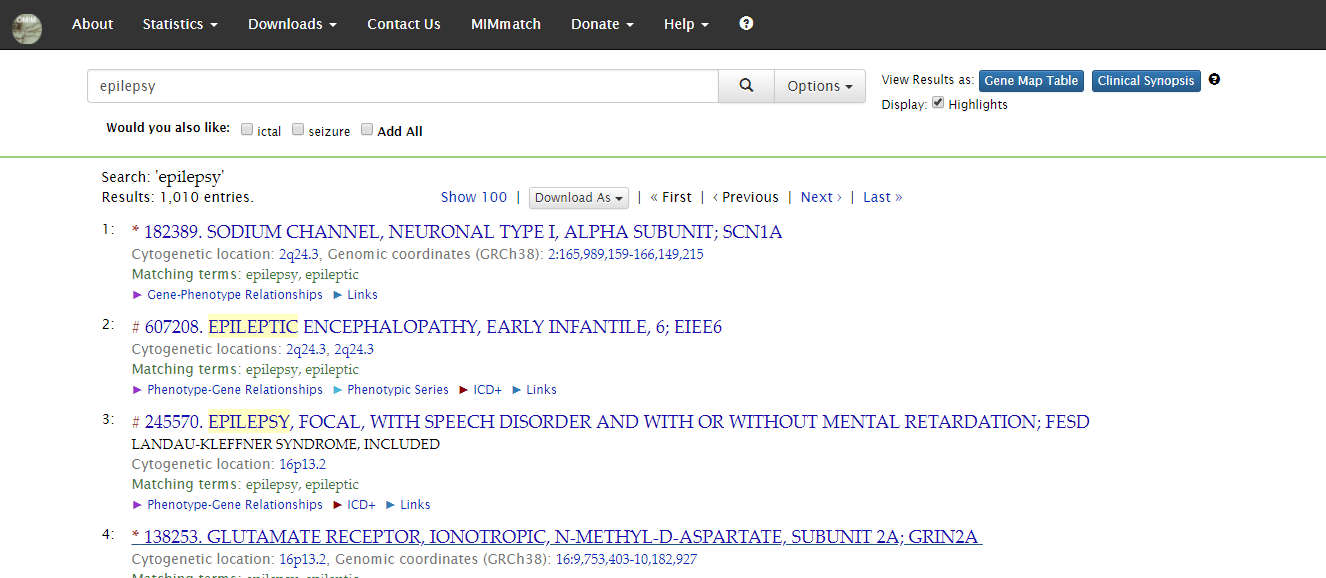
点击download as下载相关文件。下载下来的文件里,需要的是‘Phenotype MIM number’。可以把这些数字单独弄成一个文件。
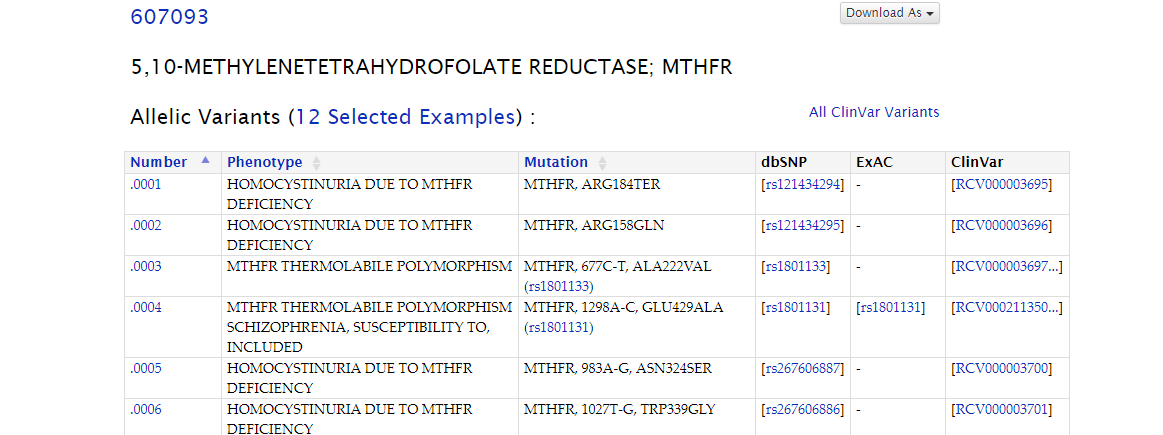
比如说,我有607093这个OMIM ID。就可以凭ID找到相关的页面。
还可以找到相关致病位点(Clinvar中提示为致病的位点)。
一开始,其实我是想用request.get()来把这个页面的源代码爬下来然后再用BeautifulSoup分析的。
然而,先不说源代码的格式很乱很麻烦,OMIM贴心的又提供了一个download按钮。 所以,可以用下面的代码批量下载这些文件。 我是直接调用默认的浏览器来下载数据,这样不会触发反爬虫机制。
import time
import random
import sys
import webbrowser
sys.path.append('libs')
# 我把所有的id放在一个文件里了
omid = open('omid.txt', 'r')
for line in omid:
om = line.split('\n')[0]
n = random.randint(1, 5)
url = 'https://www.omim.org/allelicVariant/' + om + '?format=tsv'
webbrowser.open(url, new=2)
print str(om)
# 要有时间间隔的爬数据,不然别人发现会ban掉你的IP
# 还有就是大量瞬间爬数据会让别人网站崩溃,不可以做这种事
time.sleep(5 + n)
爬下来的文件,看了看格式把它们合并起来就行了。我把这些文件都放在’all’文件夹下,用以下代码合并
import os
outputfile = open('ep_omim.txt', 'w')
outputfile.write('Number Phenotype Mutation dbSNP ExAC dbSNP ClinVar' + '\n')
for file in os.listdir('all'):
inputfile = open(os.path.join('all', file), 'r')
lines = inputfile.readlines()
count = 0
for line in lines:
count += 1
if count <= 9:
continue
else:
if line == '\n':
continue
else:
outputfile.write(line)
inputfile.close()
outputfile.close()
以上。就做出一个简单的panel了。 后续的完善需要做的工作还很多呢。