该来的还是会来的。
上一期说了,Hisat2+Stringtie+Ballgown这个流程,是文章27560171推荐的,比较快。但是因为Ballgown不是以reads数作为统计基础的,所以可能没有DESeq2这种准确。
上次下载的测试数据,是单独把chrX染色体弄出来的。 我把数据都分门别类放好了,要养成这样的习惯。
 上面的是原始数据。
首先要做的是,把原始数据和索引文件用hisat2进行比对,来生成sam文件。然后用samtools转成bam文件和排序。
注意下面的sample要改成各个sample的名字。
上面的是原始数据。
首先要做的是,把原始数据和索引文件用hisat2进行比对,来生成sam文件。然后用samtools转成bam文件和排序。
注意下面的sample要改成各个sample的名字。
# 这里-p表示线程数,-x后面跟的是索引文件的文件夹名,测试数据下载下来会有。
# 如果是完整的人类数据的话,对应的是上次下载的grch38_tran这些文件。
hisat2 -p 8 -x chrX_tran \
-1 sample_1.fastq.gz \
-2 sample_2.fastq.gz \
-S sample.sam
samtools view -bS \
sample.sam \
> sample.bam
samtools sort sample.bam \
-o sample.sort.bam
处理完是这样子的:
接下来,是用stringtie进行组装,输出gtf文件。
# 这里的-G后面的是参考的注释文件
# 对应到人类数据是Homo_sapiens.GRCh38.92.chr_patch_hapl_scaff.gtfzhege .
stringtie -p 8 -G chrX.gtf sample.sort.bam -o sample.gtf
结果展示:
下一步,是把这些gtf数据进行合并。
stringtie --merge -p 8 -G chrX.gtf \
-o stringtie_merge.gtf \
mergelist.txt
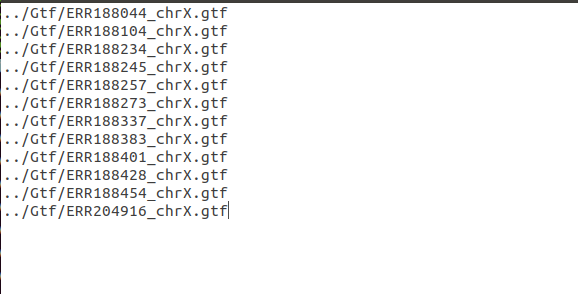
这里的mergelist.txt文件,是把每个gtf文件都写去,路径要对。比如,我的是这样的:
next,重新组装,并且为ballgown建立读入文件。
# 这里的-B命令就是输出ballgown需要的文件的。
stringtie sample.sort.bam -e -B \
-p 8 \
-G stringtie_merge.gtf \
-o Ballgown/sample/sample.gtf
然后该目录的结构大概是这样的。
最后就是跑Ballgown的流程了。注意是用R脚本。
library(ballgown)
library(RSkitterBrewer)
library(genefilter)
library(dplyr)
library(devtools)
# 设定工作区域
setwd("~/PATH to/RNA-seq")
# 读取一个状态文件,该文件的内容可以通过下载测试数据自己去看
pheno_data <- read.csv("Ballgown/geuvadis_phenodata.csv")
# 导入
bg_chrX = ballgown(dataDir="Ballgown", samplePattern="ERR", pData=pheno_data)
# 过滤
bg_chrX_filt = subset(bg_chrX, "rowVars(texpr(bg_chrX))>1", genomesubset=TRUE)
# 转录本的差异表达矩阵,增加基因名,按p值排序
transcripts_result = stattest(bg_chrX_filt, feature="transcript",
covariate="sex",
adjustvars=c("population"),
getFC=TRUE,
meas="FPKM"
)
transcripts_result = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt),
geneIDs=ballgown::geneIDs(bg_chrX_filt),
transcripts_result
)
transcripts_result = arrange(transcripts_result, pval)
# 基因的差异表达矩阵,增加基因名,按p值排序
genes_result = stattest(bg_chrX_filt, feature="gene",
covariate="sex",
adjustvars=c("population"),
getFC=TRUE,
meas="FPKM"
)
genes_result = data.frame(geneNames=ballgown::geneNames(bg_chrX_filt),
geneIDs=ballgown::geneIDs(bg_chrX_filt),
genes_result
)
genes_result = arrange(genes_result, pval)
# 输出结果
write.csv(transcripts_result, "Ballgown/chrX_transcripts_result.csv", row.names=FALSE)
write.csv(genes_result, "Ballgown/chrX_genes_result.csv", row.names=FALSE)
本期结束,其实还有画图部分,在DESeq2流程结束再一起说吧,我还没研究好。