这是官网:qualimap
这个软件就像fastqc那样,又做了命令行,又做了GUI界面。总体让我感觉非常好。 首先来下载安装。
wget https://bitbucket.org/kokonech/qualimap/downloads/qualimap_v.2.12.zip
unzip qualimap_v.2.12.zip
cd qualimap_v.2.12
./qualimap -h
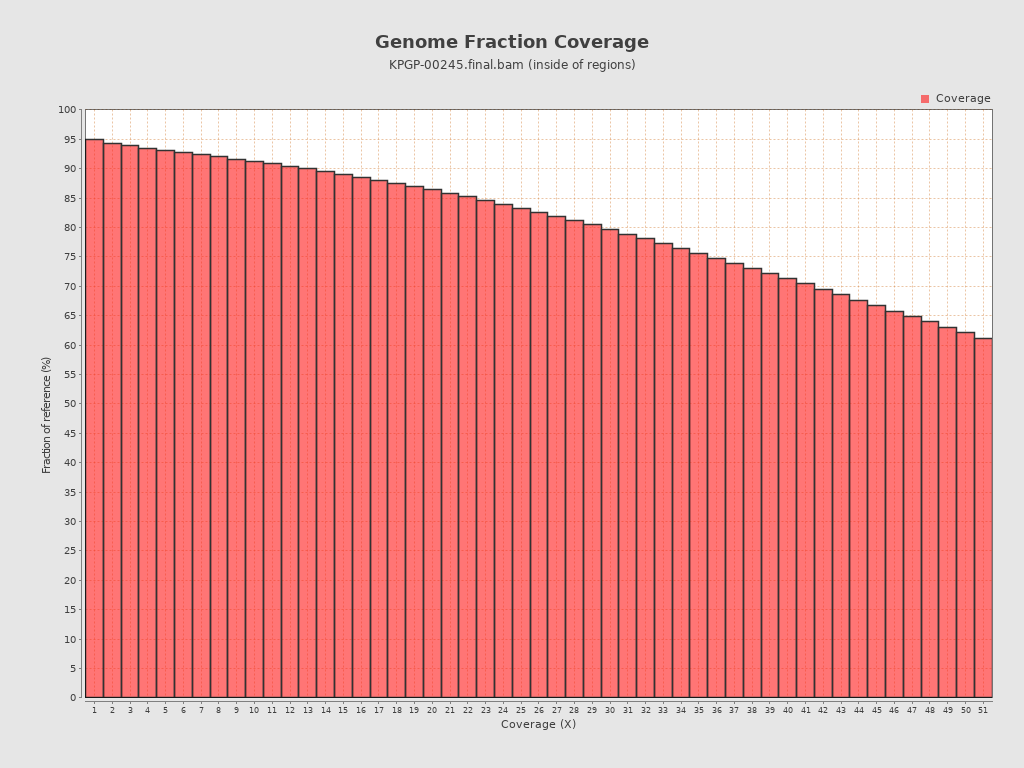
然后它可以用来统计比对之后的bam文件的数据质量,连深度覆盖度这些都可以统计。 这一点,fastqc是做不到的。 想统计fastqc还可以用samtools。不过我觉得用qualimap比较友好。连图都画好了呀!
# 主要的命令就是这样,会在同一个bam文件的文件夹下生成统计结果
qualimap bamqc -bam in.bam
我在跑上面这条命令的时候,提示我内存不足,所以我加了一个参数。
qualimap bamqc -bam in.bam --java-mem-size=8G
然后一开始我发现我的统计结果coverage图很奇怪。想了想,应该是因为我的这个全外显子是用了全基因组作为参考,然后因为有大量的区域没有任何的reads,导致coverage被拉到差不多平均为0。所以必须指定区域,就是需要一个bed文件。 然后我去找了GATK的外显子interval_list←点这个下载b37版本的。
接下来用一个python脚本把它转成bed文件格式。
a = open('Broad.human.exome.b37.interval_list', 'r')
b = open('Broad.human.exome.b37.bed', 'w')
for line in a:
if line.startswith('@'):
continue
else:
lineAS = line.split('\n')[0].split('\t')
l = [lineAS[0], lineAS[1], lineAS[2], lineAS[4], "0", lineAS[3]]
out = '\t'.join(l)
b.write(out + '\n')
b.close()
a.close()
然后重新跑,这次再加一个参数
qualimap bamqc -bam in.bam --java-mem-size=8G -gff Broad.human.exome.b37.bed
结果看起来不错!