感觉上如果做扩增子的东西始终要懂怎么用qiime2。。。 qiime2把每一步的文件都封装成qza文件,然后画出来的图都封装成qzv文件。 qzv文件要到qiime view上面看。
真香警告!
之前说过怎么安装了。
启动!
docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2018.6
导入数据
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path sample.list \
--output-path /data/Qza/paired-end-demux.qza \
--source-format PairedEndFastqManifestPhred33
sample.list长这样:
sample-id,absolute-filepath,direction
sample-1,$PWD/some/filepath/sample1_R1.fastq.gz,forward
sample-2,$PWD/some/filepath/sample2_R1.fastq.gz,forward
sample-1,$PWD/some/filepath/sample1_R2.fastq.gz,reverse
sample-2,$PWD/some/filepath/sample2_R2.fastq.gz,reverse
看看情况
qiime demux summarize \
--i-data /data/demux/paired-end-demux.qza \
--o-visualization /data/demux/paired-end-demux.qzv
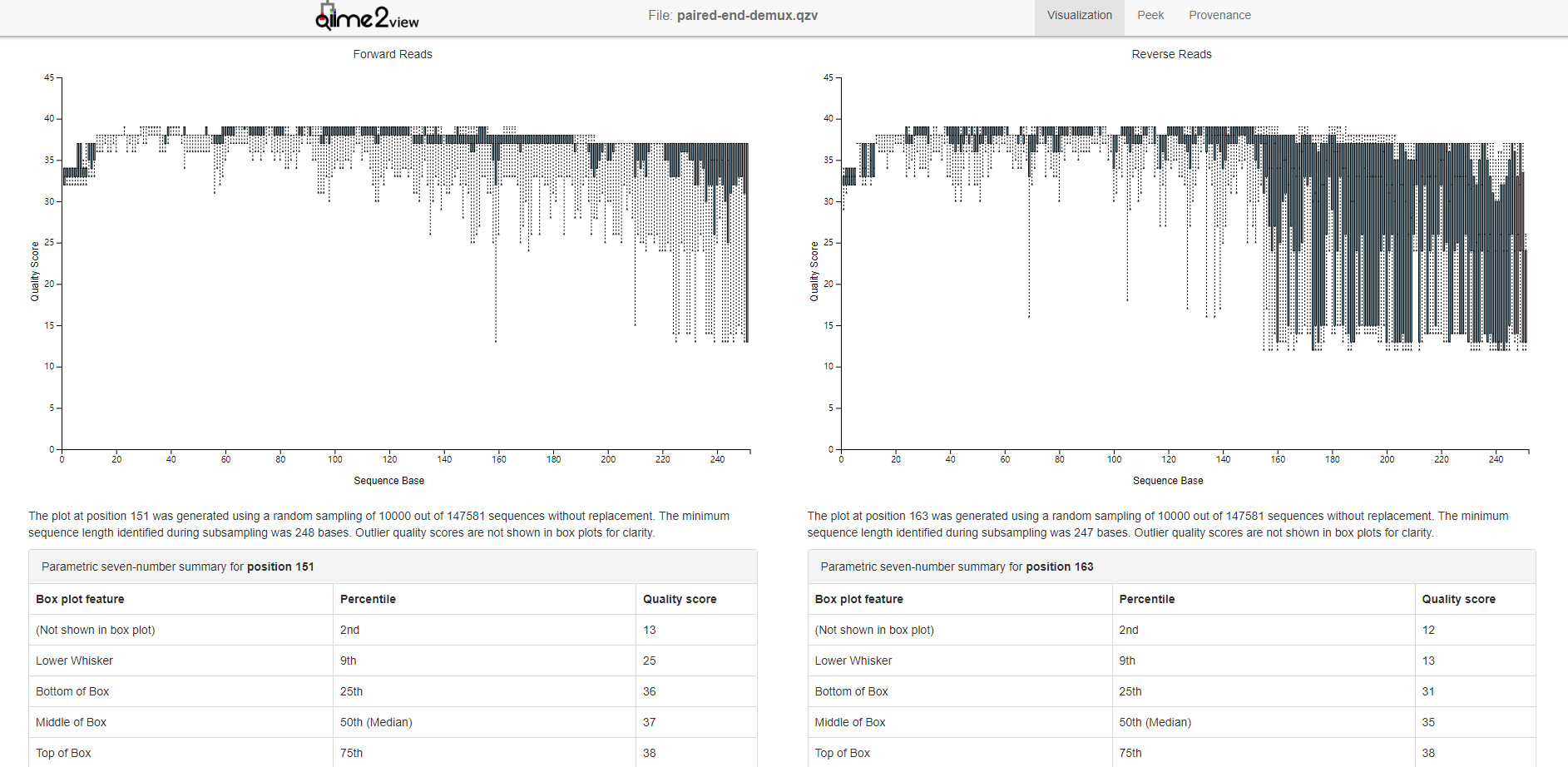
使用dada2进行分析
# 切多少得根据具体结果来呀
qiime dada2 denoise-paired \
--i-demultiplexed-seqs /data/demux/paired-end-demux.qza \
--p-trunc-len-f 240 \
--p-trunc-len-r 160 \
--o-representative-sequences /data/dada2/rep-seqs-dada2.qza \
--o-table /data/dada2/table-dada2.qza \
--output-dir ./temp \
--p-n-threads 8
看看情况
qiime feature-table summatize \
--i-table /data/dada2/table-dada2.qza \
--o-visualization /data/dada2/table.dada2.qzv
# 输出一个biom文件
qiime tools export \
/data/dada2/table.dada2.qza \
--output-dir exported-data
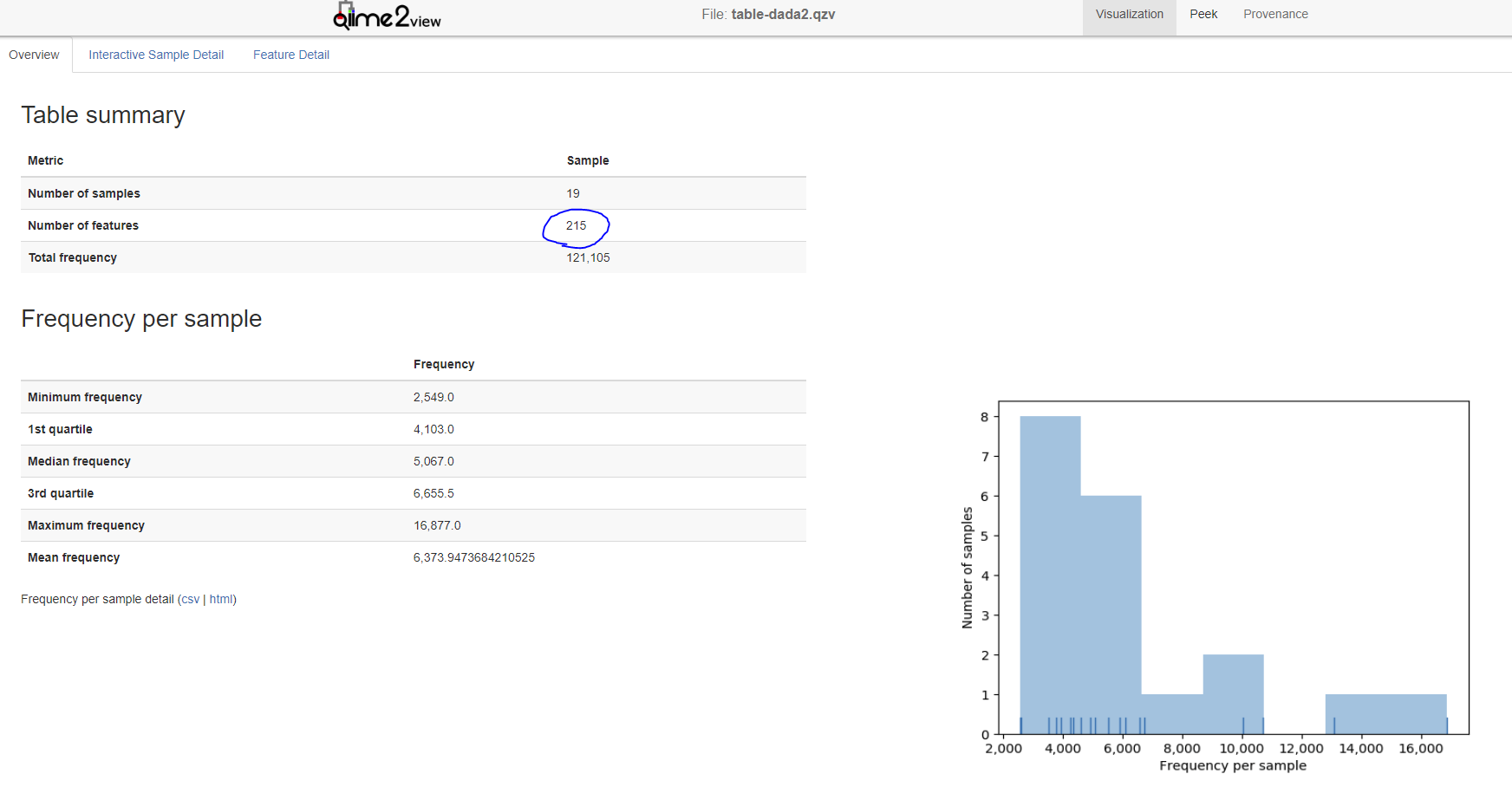 19个样本检测到215种微生物,和之前单纯的dada2流程一致。
19个样本检测到215种微生物,和之前单纯的dada2流程一致。
构建系统发育树
qiime alignment mafft \
--i-sequences /data/dada2/rep-seqs-dada2.qza \
--o-alignment /data/tree/aligned-rep-seqs.qza
qiime alignment mask \
--i-alignment /data/tree/aligned-rep-seqs.qza \
--o-masked-alignment /data/tree/masked-aligned-rep-seqs.qza
qiime phylogeny fasttree \
--i-alignment /data/tree/masked-aligned-rep-seqs.qza \
--o-tree /data/tree/unrooted-tree.qza
qiime phylogeny midpoint-root \
--i-tree /data/tree/unrooted-tree.qza \
--o-rooted-tree /data/tree/rooted-tree.qza
alpha分析和beta分析
这里的sampling-depth,可以去看table.dada2.qzv来决定。具体是选择一个值来过滤掉未达标的样本。 sample-metadata.txt是一个记录样本信息的文件,可以把参考信息都写进去。第一列必须是sampleid。后面用tab分割。 每一列是一个信息。比如可以加入otu序列或者检测时间,受检者性别等等等等的信息。其实就是分类信息。比较自由。
qiime diversity core-metrics-phylogenetic \
--i-phylogeny /data/tree/rooted-tree.qza \
--i-table /data/dada2/table-dada2.qza \
--p-sampling-depth 2500 \
--m-metadata-file sample-metadata.txt \
--output-dir core-metrics
输出是一堆emperor的成分分析图。 alpha:faith图和evenness图,这两个图是需要metadata里有连接的引物序列的,所有这里不画了。
qiime diversity alpha-group-significance \
--i-alpha-diversity /data/core-metrics/faith_pd_vector.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization /data/core-metrics/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity /data/core-metrics/evenness_vector.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization /data/core-metrics/evenness-group-significance.qzv
beta分析 可以通过改变–m-metadata-column来选择要输出的信息类型,这取决于metadata中的列名。
qiime diversity beta-group-significance \
--i-distance-matrix /data/core-metrics/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.txt \
--m-metadata-column dpw \
--o-visualization /data/core-metrics/unweighted-unifrac-time-site-significance.qzv \
--p-pairwise
emperor画PCoA图
qiime emperor plot \
--i-pcoa /data/core-metrics/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.txt \
--p-custom-axes DaysSinceExperimentStart \
--o-visualization /data/core-metrics/unweighted-unifrac-emperor-DaysSinceExperimentStart.qzv
qiime emperor plot \
--i-pcoa /data/core-metrics/bray_curtis_pcoa_results.qza \
--m-metadata-file sample-metadata.txt \
--p-custom-axes DaysSinceExperimentStart \
--o-visualization /data/core-metrics/bray-curtis-emperor-DaysSinceExperimentStart.qzv
alpha分析:
qiime diversity alpha-rarefaction \
--i-table /data/dada2/table.dada2.qza \
--i-phylogeny /data/tree/rooted-tree.qza \
--p-max-depth 25000 \
--m-metadata-file sample-metadata.txt \
--o-visualization /data/core-metrics/alpha-rarefaction.qzv
物种分类注释
不知道为啥qiime2要把分类注释放这么后来解释。。。 既然是分类注释,首先肯定得有个现成的数据库。qiime2可以用来训练自己的数据库,也可以使用现成训练好的。 根据实际情况和不同使用场景自行选择就好了。 点击进入自行训练的教程。 点击进入现成的数据库。 现成的数据库说了一堆注意事项,还标红了,好可怕!一定要谨慎使用。 这里还是用silva 132版本的。 这里居然!需要!20G以上的内存(RAM)才能跑!不然的话就会报memory error!我的电脑才16G内存!看来要放弃这一步了。
qiime feature-classifier classify-sklearn \
--i-classifier silva-132-99-515-806-nb-classifier.qza \
--i-reads /data/dada2/rep-seqs-dada2.qza \
--o-classification /data/taxa/taxonomy.qza
qiime metadata tabulate \
--m-input-file /data/taxa/taxonomy.qza \
--o-visualization /data/taxa/taxonomy.qzv
qiime taxa barplot \
--i-table /data/dada2/table-dada2.qza \
--i-taxonomy /data/taxa/taxonomy.qza \
--m-metadata-file sample-metadata.txt \
--o-visualization /data/taxa/taxa-bar-plots.qzv
只能放张示意图了。
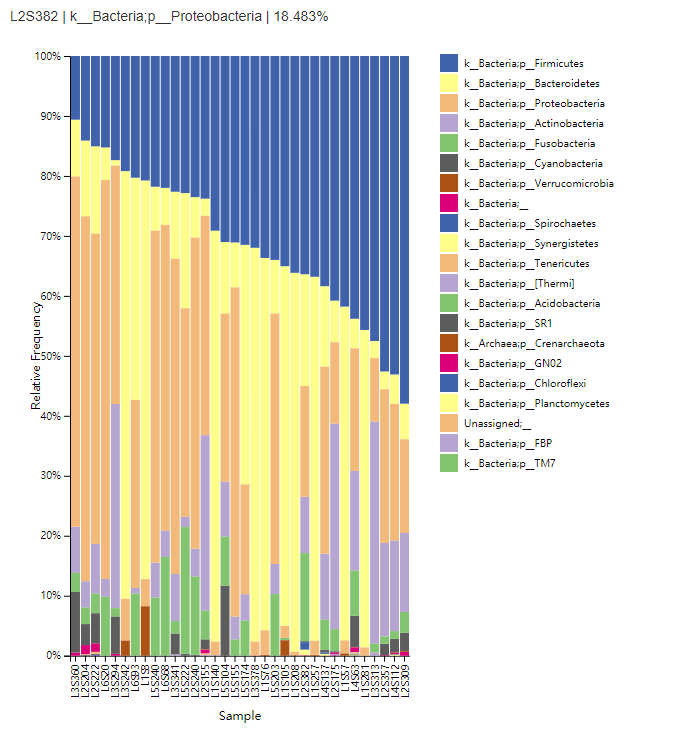
这张图是qiime2输出的选择以门为分类组别的丰度图。
总结一下。我个人觉得,如果产品面向的是消费者的级别,直接用R包dada2这样做,比较好。 主要在于自动化的起来比较容易,而且对资源不太吃紧。而qiime2适合用于研发项目,因为它能提供更多的参数。 各种工具的高度集中。另外由于qiime2的作图格式都是qzv,需要通过浏览器等来查看,这一点也不利于自动化(我还没找到qiime自动出图的方法,不一定是没有)。
ps. 其实可以参考qiime2整合的工具,从中挑选出需要的,再去通过单独安装这些工具来实现自动化。
pss. qiime2给我的感觉是各种参数设置其实让人挺舒服的,–i、–o这样的。
psss. 目前我个人觉得做比较简单的一套分析流程,就是数据过滤→聚类/降噪→注释→物种丰度→alpha/beta→主成分分析→差异分析。