这一篇文章将会说一下ChIP-seq的基本流程,以及我自己的一些理解(不一定是对的!),主要参考的是生信技能树jimmy的教程。
基本知识(个人理解)
chip-seq的原理首先是把蛋白质和DNA交联(cross-linking),然后利用超声波打散成片段(sonication),再利用抗原抗体特异识别把目的蛋白的DNA片段沉淀下来再反交联得到DNA,最后就是送出测序。
一般的,还需要有阴性对照,可以用经过超声波打断但是没有进行免疫沉淀的总DNA作为对照,也可以用igG作为对照,因为igG理论上不会免疫沉淀(应该是这样?)。
然后得到了测序结果,比对到参考基因组,然后用阴性对照作为去除背景噪音的材料,就可以得到实验组的DNA的富集峰(就是那里测到特别多,我是这样理解的)。用软件把这些富集峰(peaks)都call出来,由于有了参考,可以得到位置信息和丰度。再来就是用数据库进行注释,就是注释出这些peak都落在什么基因上,都在哪些位置。
因此,ChIP-seq可以解决的问题是蛋白的DNA在基因组的什么位置、基因表达是否由这种(或者多种)复合物来调控、多种复合物是否共同发挥作用,总结起来就是用来研究DNA和蛋白的相互作用。
数据下载
用的数据是GEO:GSE42466;BioProject:PRJNA182214。
参考文献:23273917
我个人比较喜欢直接在EMBL下载数据,这样直接下下来fq文件,不需要sratk慢慢解压了。
for ((i=204; i<=208; i++)); \
do wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR620/SRR620$i/SRR620$i.fastq.gz; \
done
下载了5个样本,分布是Ring1B、cbx7、SUZ12、RYBP、IgGold。
下载参考基因组,这是小鼠的基因组
wget http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz
tar -zxvf chromFa.tar.gz
# 把每条染色体的fasta合并起来
cat *.fa > ucsc.mm10.fa
软件和R包安装
这次用到的软件主要有比对的Bowtie2、peak calling的MACS2、以及用来做注释的Y叔写的R包ChIPseeker。 具体的安装流程记不太清,凭记忆写的!实际操作的时候自己看着来!应该不会差太多!
Bowtie2
wget https://github.com/BenLangmead/bowtie2/releases/download/v2.3.4.2/bowtie2-2.3.4.2-linux-x86_64.zip
unzip bowtie2-2.3.4.2-linux-x86_64.zip
MACS2
wget https://github.com/taoliu/MACS/archive/2015.4.20.tar.gz
tar -zxvf 2015.4.20.tar.gz
cd 2015.4.20
sudo python setup.py install
注意这里有坑! MACS2需要依赖Cython才能跑,所以还得先装Cython
sudo apt-get install python-dev
wget https://github.com/cython/cython/archive/0.28.5.tar.gz
tar -zxvf 0.28.5.tar.gz
cd 0.28.5
sudo python setup.py install
然后再MACS2的文件夹中
sudo python setup_w_cython.py install
cd bin
chmod +x macs2
./macs2 -h
这样才装好。
R包ChIPseeker
source("https://bioconductor.biocLite.R")
biocLite("ChIPseeker")
我在windows下是这样就装好了,可是在ubuntu下会报错,说是一个依赖的包RMySQL没装上。 然后看了一下这个包没装上是因为ubuntu里没装mysql的client端。 所以是这样解决的,先在R中装好DBI
# install.packages("devtools")
library("devtools")
devtools::install_github("r-dbi/DBI")
然后在shell中
sudo apt-get install mysql-client
sudo apt-get install libdbd-mysql
sudo apt-get install libmysqlclient-dev
wget https://cran.r-project.org/src/contrib/RMySQL_0.10.15.tar.gz
R CMD INSTALL --configure-args='--with-mysql-dir=/usr/lib/mysql' RMySQL_0.10.15.tar.gz
再来安装ChIPseeker就可以了
source("https://bioconductor.biocLite.R")
biocLite("ChIPseeker")
质控
简单的用fastqc和multiqc看看测序结果
fastqc Rawdata/*.fastq.gz -o ./QC
multiqc ./QC
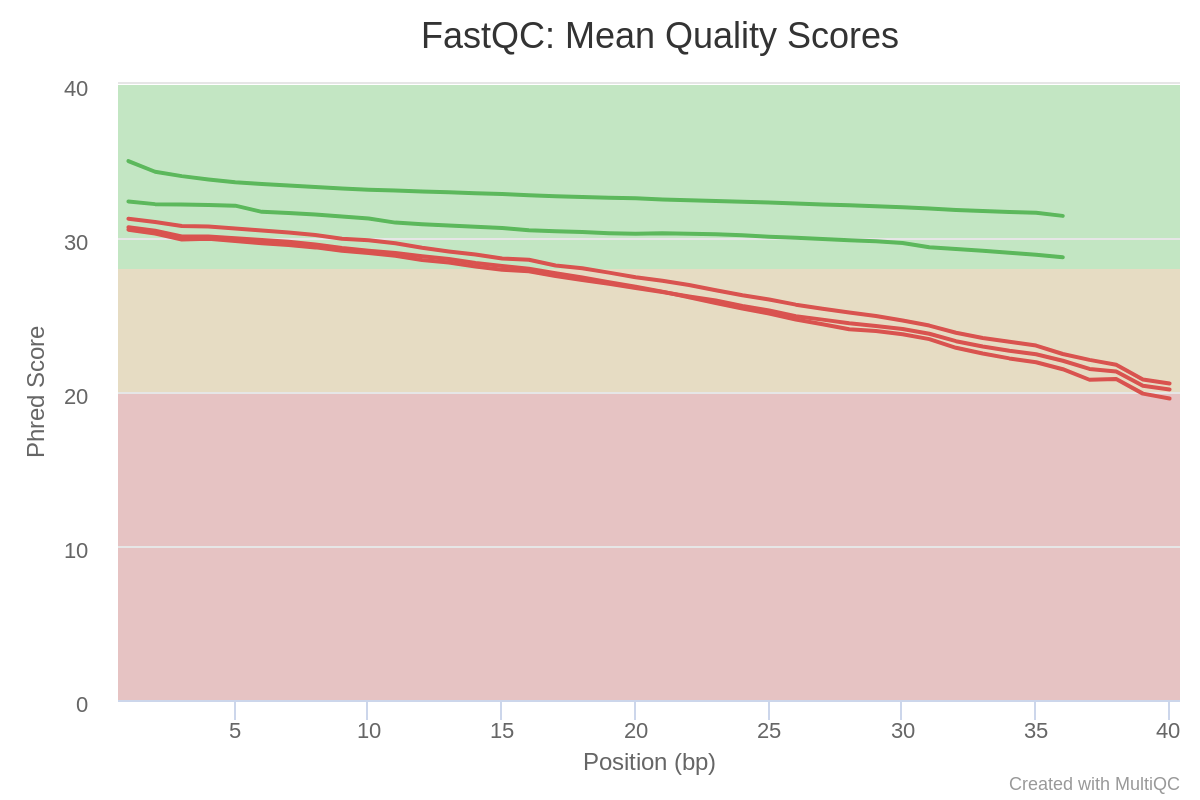
看图,明显的看出Ring1B、SUZ12、IgGold这三个样本的3’端质量不太好,为了和另外一个样本保持一致,可以用软件剪掉。由于Bowtie2带有这种功能,所以这里先不用别的软件了,可以在比对的时候剪掉5个bp。
比对
先对参考基因组建立索引
bowtie2-build -p 8 ucsc.mm10.fa ucsc.mm10
接下来进行比对,我的原始数据是放在Rawdata文件夹下而且已经用样本名来重命名了,可以这样批量运行。
filenames=$(ls Rawdata/*.fastq.gz)
for file in $filenames;do
sample=$(basename ${file} .fastq.gz)
bowtie2 -p 8 -x ucsc.mm10 \
-3 5 --local \
-U Rawdata/${sample}.fastq.gz \
| samtools sort -@ 8 -O bam \
-o Bam/${sample}.sorted.bam
samtools index Bam/${sample}.sorted.bam
done
然而,之前说了,其实只有三个样本需要切5个bp的碱基,上面这样实际上是把所有的样本都切了,所以我还是逐个来运行了。。。
要切的就这样,举个例子,这里就是说把3’切5个bp。
sample=Ring1B
bowtie2 -p 8 -x ucsc.mm10 \
-3 5 --local \
-U Rawdata/${sample}.fastq.gz \
| samtools sort -@ 8 -O bam \
-o Bam/${sample}.sorted.bam
samtools index Bam/${sample}.sorted.bam
不需要的,就把参数删掉就好了
sample=cbx7
bowtie2 -p 8 -x ucsc.mm10 \
-U Rawdata/${sample}.fastq.gz \
| samtools sort -@ 8 -O bam \
-o Bam/${sample}.sorted.bam
samtools index Bam/${sample}.sorted.bam
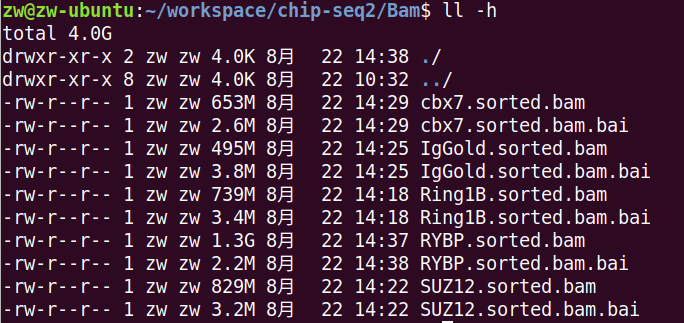
结果像这样。
Peaks Calling
接下来就可以对这些bam文件,用macs2进行peaks calling了。
macs2 callpeak \
-c IgG.sorted.bam \
-t cbx7.sorted.bam \
-q 0.05 \
-f BAM \
-g mm \
-n cbx7 \
--outdir ../Peak
这里是其中一个例子,其他样本也按照这个来跑。其中-c是control组,一般就是IgG阴性对照,然后-t是treatment组。 -g参数要输入的是基因组的大小,但是macs2预设了几个选择,比如人类的hs,比如小鼠的mm。
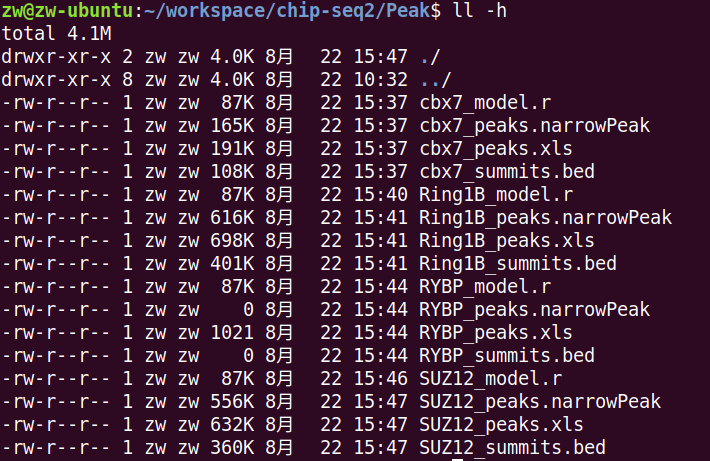
结果如图,明显看出RYBP这个样本有问题,其实跑过这个数据的大佬们都提到过了,估计是作者上传数据的时候传错了。 所以这里可以去下载作者上传的peaks。
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42466/suppl/GSE42466_Cbx7_peaks_10.txt.gz
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42466/suppl/GSE42466_RYBP_peaks_5.txt.gz
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42466/suppl/GSE42466_Ring1b_peaks_10.txt.gz
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42466/suppl/GSE42466_Suz12_peaks_10.txt.gz
但是由于现在只是为了流程式的演示ChIP-seq,所以我也不打算用作者提供的peak。直接去除RYBP这个样本,用剩下的3个样本来进行下面的操作。
Peak基本信息
现在就是用ChIPseeker这个R包了。参考的是Bioconductor上的ChIPseeker文档。
首先是加载各种包,需要注意的是,txdb需要在clusterProfiler前读入,如果先加载了clusterProfiler再读入txdb,会出现错误。当然,如果不做差异分析,也就没必要加载clusterProfiler。
library(ChIPseeker)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
library(clusterProfiler)
library(org.Mm.eg.db)
library(ReactomePA)
txdb其实就是注释的数据库,也可以自行创建。一般的,可以去UCSC Table Browser中找自己想要的下载。 这里是直接使用了预设的mm10注释库了。
接下来是把peak读入
cbx7 <- readPeakFile("Peak/cbx7_peaks.narrowPeak", header=FALSE)
Ring1B <- readPeakFile("Peak/Ring1B_peaks.narrowPeak", header=FALSE)
SUZ12 <- readPeakFile("Peak/SUZ12_peaks.narrowPeak", header=FALSE)
# 把样本都放一个列表里,之后有用
peak_list <- list(cbx7=cbx7, Ring1B=Ring1B, SUZ12=SUZ12)
这里需要加个header=FALSE的参数,不然会读错。
查看peaks在基因组中的位置
covplot(cbx7, weightCol="V5")
covplot(Ring1B, weightCol="V5")
covplot(SUZ12, weightCol="V5")
# 可以单独查看某些染色体和区域
covplot(cbx7, weightCol="V5", chrs=c("chr17", "chr18"), xlim=c(4.5e7, 5e7))

示意图是cbx7的。
与TSS区域结合的peaks的概况
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrix <- lapply(peak_list, getTagMatrix, windows=promoter)
tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color=NULL)
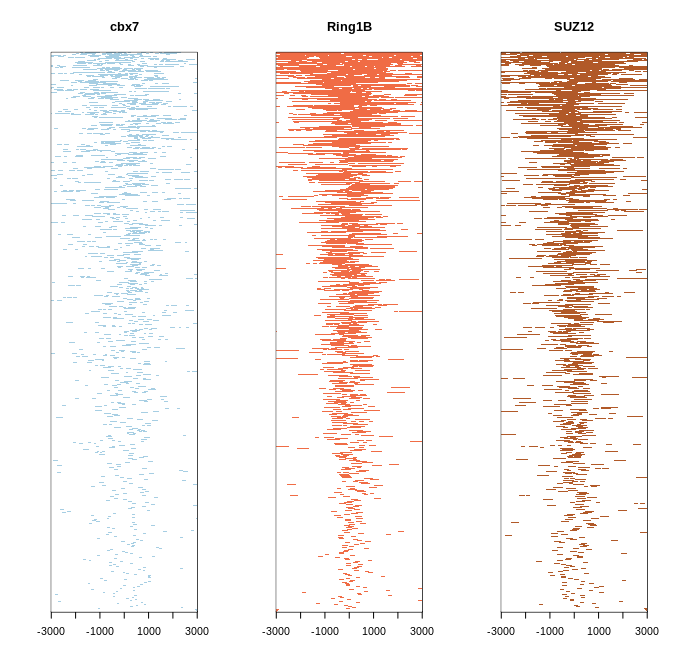
这是热图。
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency", facet="row")
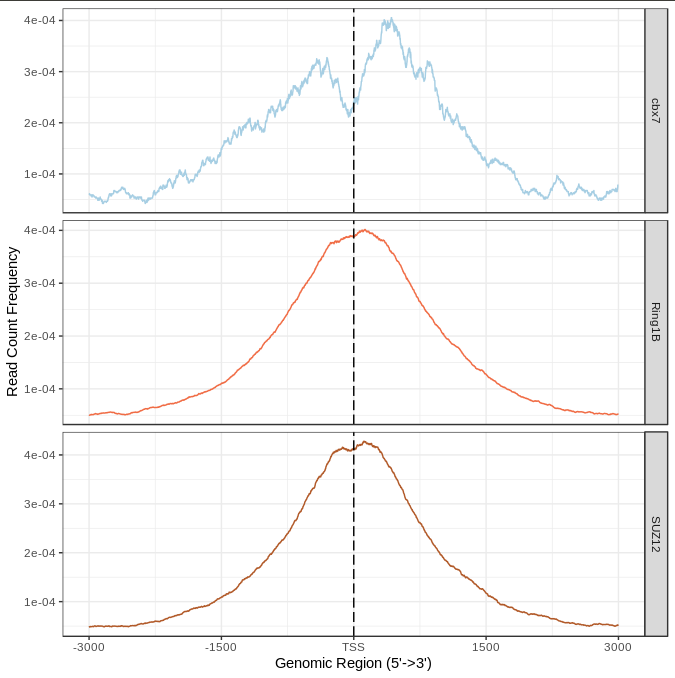
还是这种图感觉比较好看。TSS是转录起始位点,然后这里的横坐标是转录起始位点的上下3000个bp。因为一般认为,转录起始位点附近的peak的意义比较重要。
可以把三个样本合起来看,把参数里的facet去掉就行了
plotAvgProf(tagMatrix, xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")
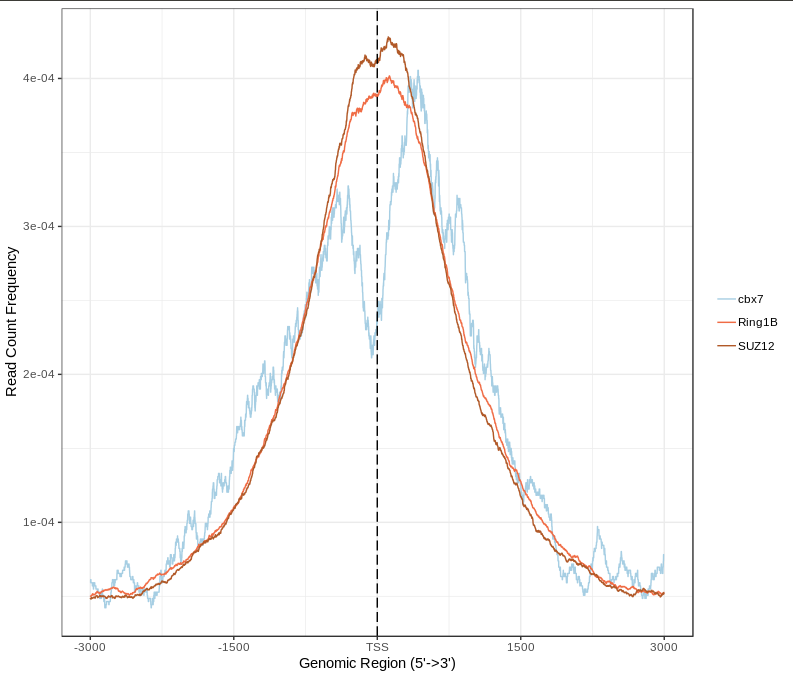
peak注释
接下来这里才是真的注释
cbx7_Anno <- annotatePeak(cbx7, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db", verbose=FALSE)
write.table(as.data.frame(cbx7_Anno), "Anno/cbx7.anno.xls", quote=F, row.names=F, sep="\t")
Ring1B_Anno <- annotatePeak(Ring1B, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db", verbose=FALSE)
write.table(as.data.frame(Ring1B_Anno), "Anno/Ring1B.anno.xls", quote=F, row.names=F, sep="\t")
SUZ12_Anno <- annotatePeak(SUZ12, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db", verbose=FALSE)
write.table(as.data.frame(SUZ12_Anno), "Anno/SUZ12.anno.xls", quote=F, row.names=F, sep="\t")
anno_list <- lapply(peak_list, annotatePeak, TxDb=txdb, tssRegion=c(-3000, 3000), verbose=FALSE, annoDb="org.Mm.eg.db")
我这里是把这些注释了然后把结果输出成表格。后面是多样本的组合。
peak可视化
单个样本可以画下面这几个图,以cbx7作为例子
plotAnnoPie(cbx7_Anno)
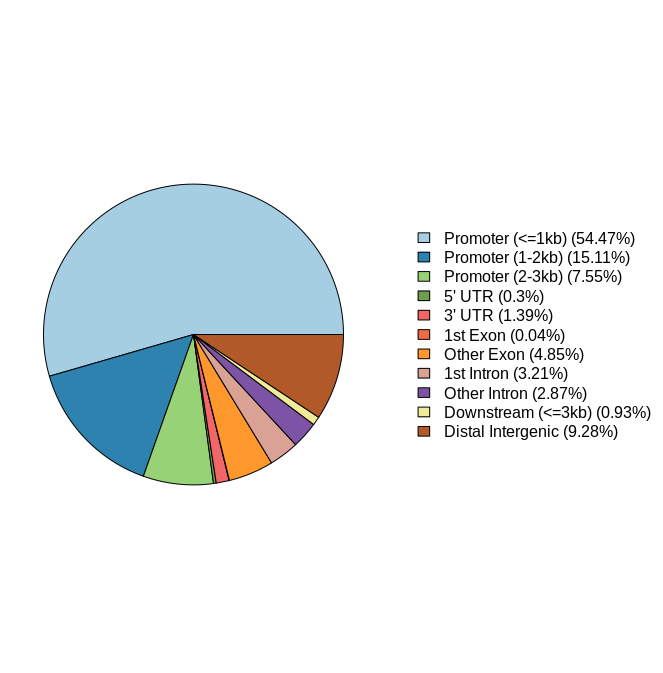
vennpie(cbx7_Anno)
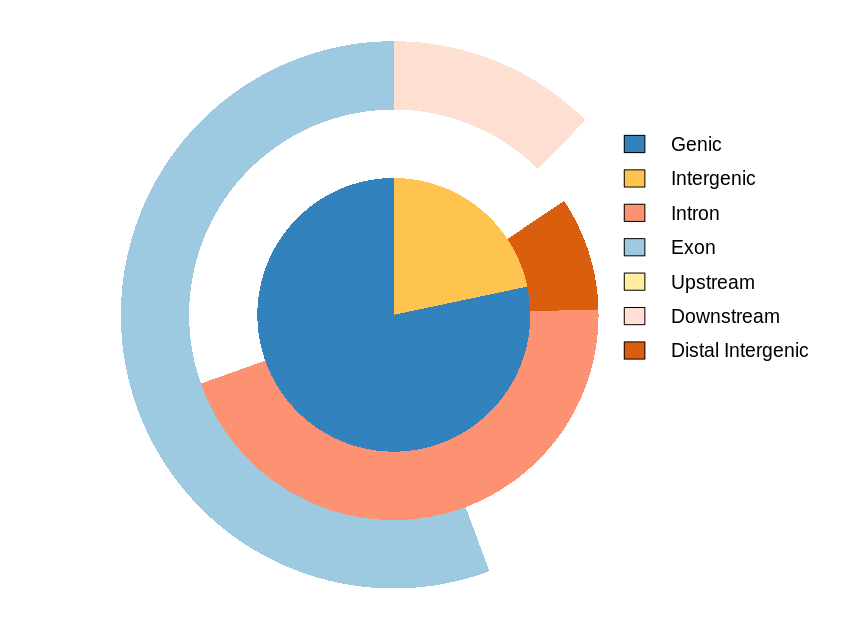
upsetplot(cbx7_Anno)
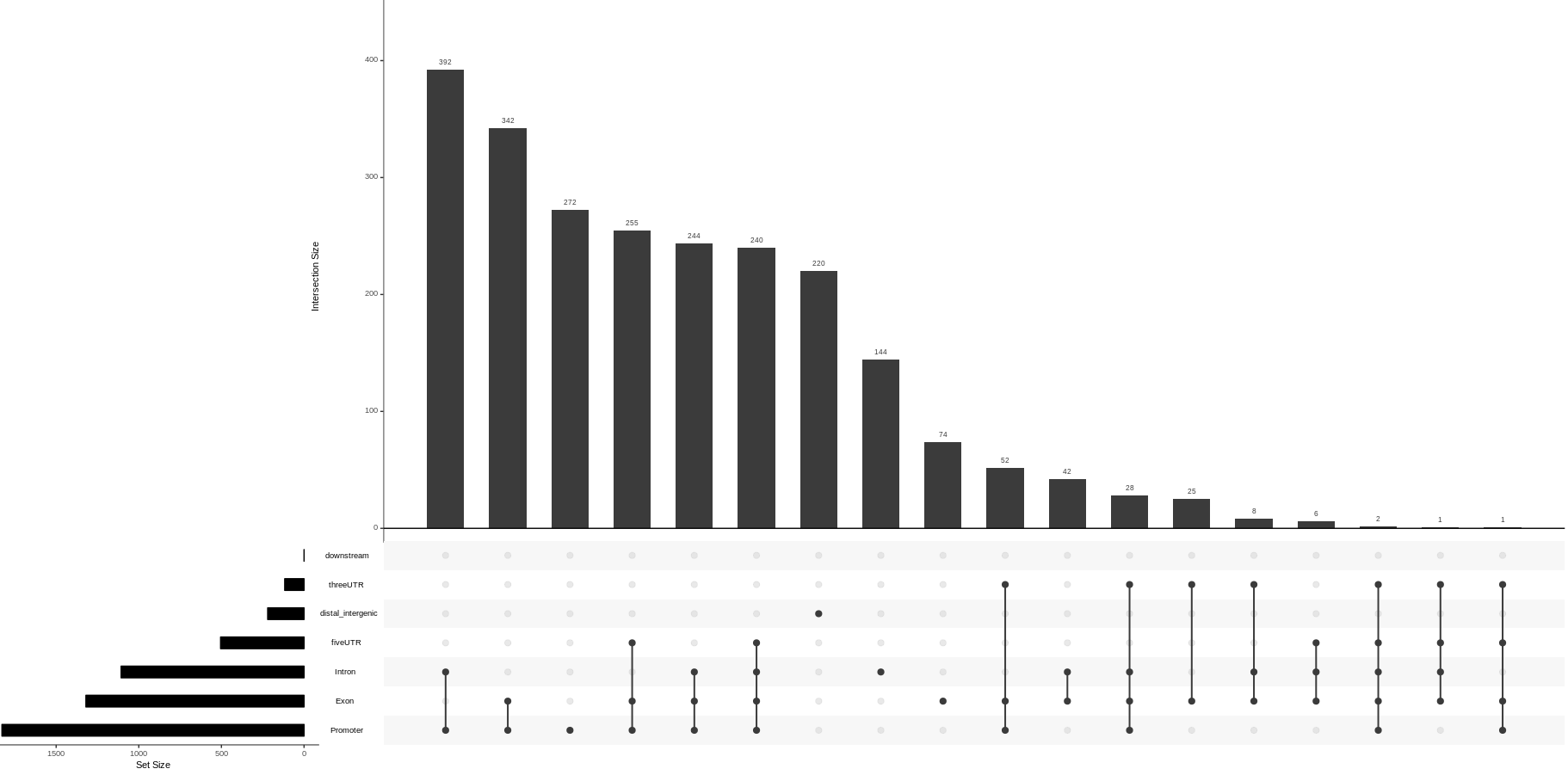
然后多个样本可以这样画(其实这些单样本也可以!)
plotAnnoBar(anno_list)
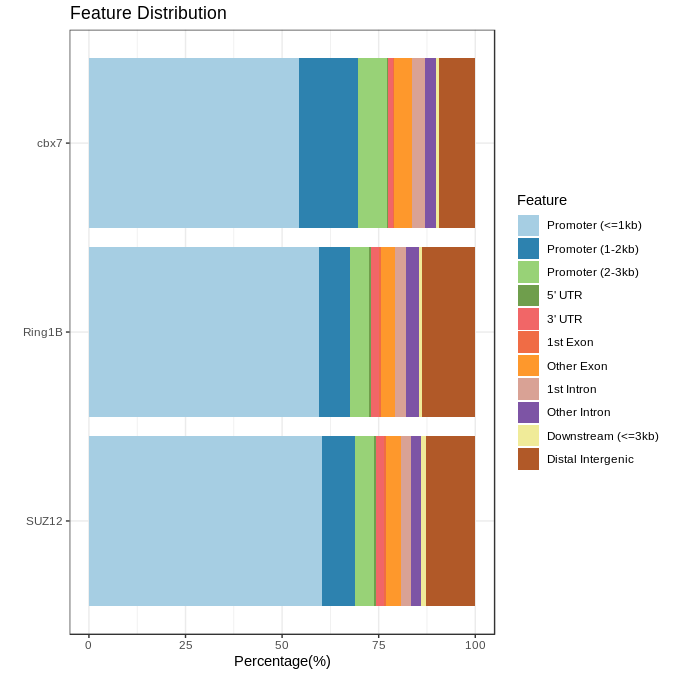
这个图实际上就是上面饼图的条形形式。
plotDistToTSS(anno_list,
title="Distribution of transcription factor-binding loci\nrelative to TSS")
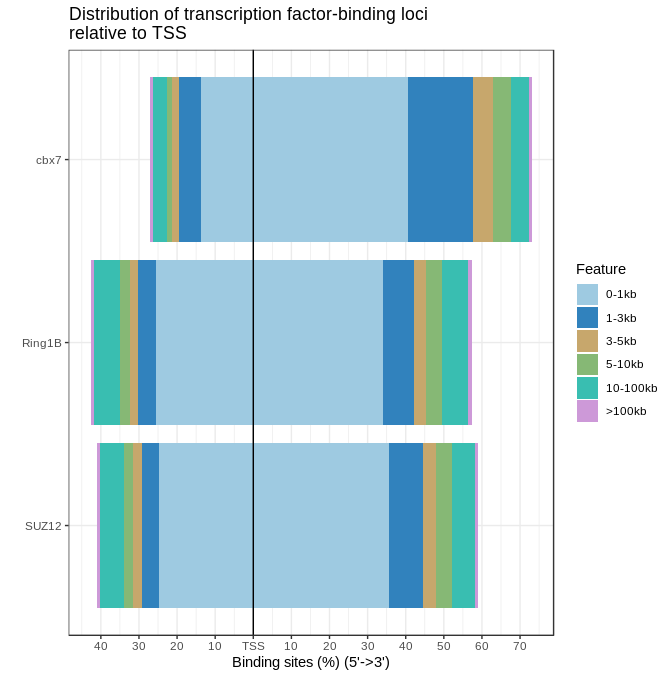
这个是TF结合基因座相对于TSS的分布(我还没搞懂这个意思。。)
富集分析
实际上ChIPseeker文档里也提供了富集分析的方法,但是我运行的时候提示这三个样本的基因并没有富集出什么通路,因此这里只是作为一个存档参考。
genes = lapply(anno_list, function(i) as.data.frame(i)$geneId)
names(genes) = sub("_", "\n", names(genes))
compKEGG <- compareCluster(geneCluster=genes, fun="enrichKEGG", pvalueCutoff=0.05, pAdjustMethod="BH")
dotplot(compKEGG, showCategory=15, title="KEGG Pathway Enrichment Analysis")
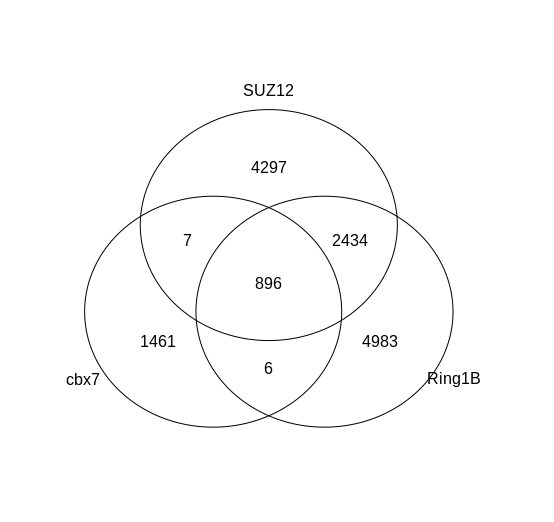
Venn图
最后画一个韦恩图,结束这篇文章。
genes = lapply(anno_list, function(i) as.data.frame(i)$geneId)
vennplot(genes)