bedtools 也是一个常驻我环境变量的软件了。
突然有一个需求,是统计任意深度的百分比,一时半会,没想到有什么现成的工具可以用,所以就用bedtools来统计了。 操作很简单,需要的输入文件是一个排过序的bam。
看一下统计原理:
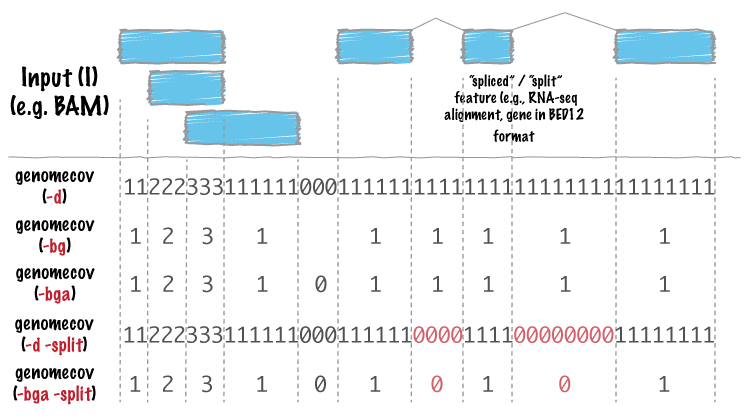
可以看出,当加入参数-d -split的时候,可以得到比较好进行下一步统计的结果。
bedtools genomecov -ibam xxx.sorted.bam -g reference.fa -bga > results.txt
例如: 得到下面这样的结果
chr1 100 101 300
chr1 101 102 311
chr1 102 150 400
chr1 150 155 320
可以看出,当加入的参数是-bga时,bedtools genomecov 输出的结果是染色体,区域起始,区域结束,这个区域的深度。
这时候就可以用python脚本来统计了,下面这个方法可以统计的是某染色体的覆盖度,染色体覆盖区域的平均深度以及染色体总体的平均深度。如果说要统计每个染色体的平均深度和覆盖度时,比较有用。
def getChromCoverage(inputfile, chrom):
inputFile = open(inputfile, "r")
length = 0
covLength = 0
counts = 0
for line in inputFile:
lineAS = line.split("\t")
chromosome = lineAS[0]
start = lineAS[1]
end = lineAS[2]
count = lineAS[3].split("\n")[0]
if chromosome == chrom:
length_tmp = int(end) - int(start)
length += length_tmp
counts += (int(count) * length_tmp)
if count != "0":
covLength_tmp = length_tmp
covLength += covLength_tmp
coverage = "%.2f" % ((float(covLength) / float(length)) * 100) + "%"
depth_cov = "%.2f" % (float(counts) / float(covLength))
depth_all = "%.2f" % (float(counts) / float(length))
inputFile.close()
return [coverage, depth_cov, depth_all]
如果是统计大于任意深度的百分比
def getDepthContent(inputfile, depth):
inputFile = open(inputfile, "r")
covLength = 0
depthLength = 0
for line in inputFile:
lineAS = line.split("\t")
chromosome = lineAS[0]
start = lineAS[1]
end = lineAS[2]
count = lineAS[3].split("\n")[0]
if count != "0":
covLength_tmp = int(end) - int(start)
covLength += covLength_tmp
if int(count) >= depth:
depthLength_tmp = int(end) - int(start)
depthLength += depthLength_tmp
covDepthPercent = "%.2f" % (float(depthLength) / float(covLength) * 100) + "%"
return covDepthPercent