以BRCA1为例,分别从三大数据库出发写几个低通量的方法。
一、NCBI
在NCBI中选择Gene,搜索BRCA1,然后点进需要的物种,先看方向,这里是反向的。然后点击下面的GeneBank,如图所示。
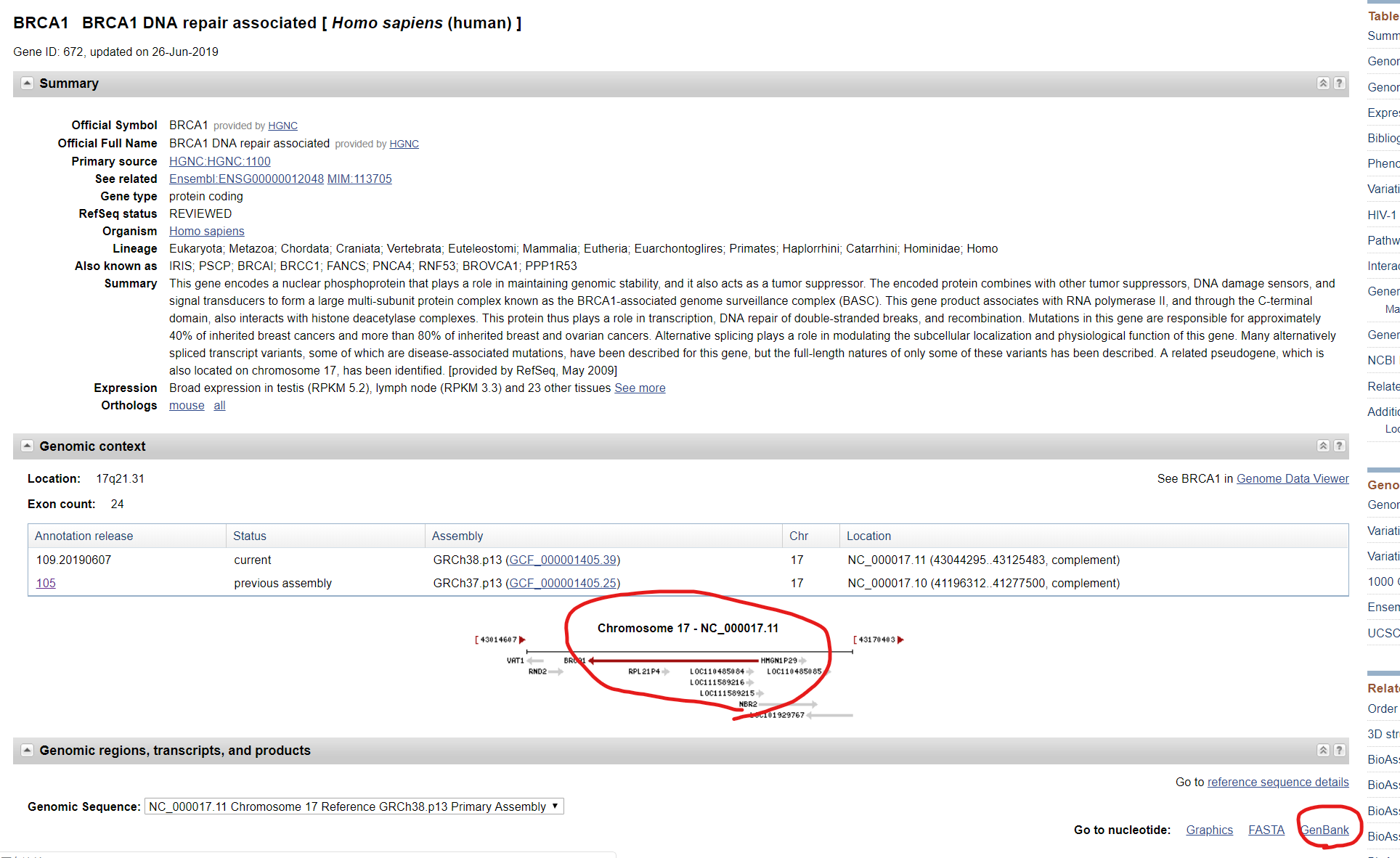
在FEATURES的mRNA这里,就可以看到每个外显子的区域。由于BRCA1的方向是反向的,所以最后面的一个才是一号外显子。外显子与外显子的中间区域就是内含子了。
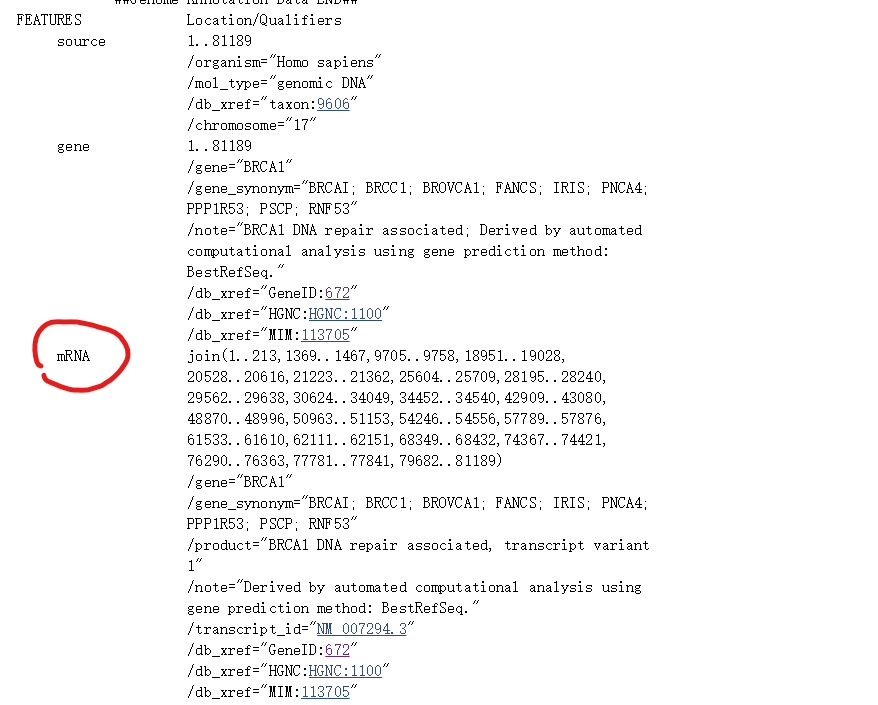
这个方法的一个比较坑的地方是,不是每个基因都有这个信息。其次,还得自己从参考基因组位置里加回去这个转录本位置才是基因组上的位置。
二、ENSEMBL
使用的是ENSEMBL的biomart,biomart有R包,可以编写程序来高通量的做这个找外显子的事情。
首先选择好DataSet,然后在Filters里打开Gene,按图里这样设置。
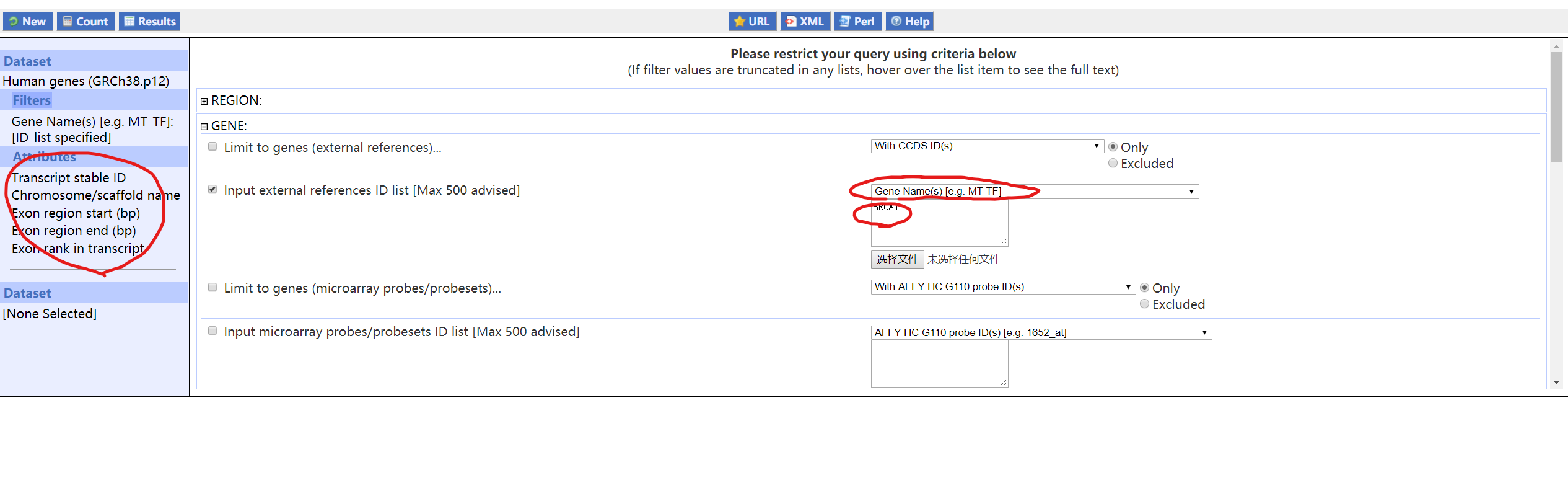
Attributes里选择Structures,我一般勾选GENE里的Chromosome/scaffold name,还有EXON里的Transcript stable ID、Exon region start (bp)、Exon region end (bp)、Exon rank in transcript。然后点击Results,就可以得到结果。
重点看同一转录本下的外显子坐标以及排序。ENSEMBL的转录本外显子大多时候都是最多的。
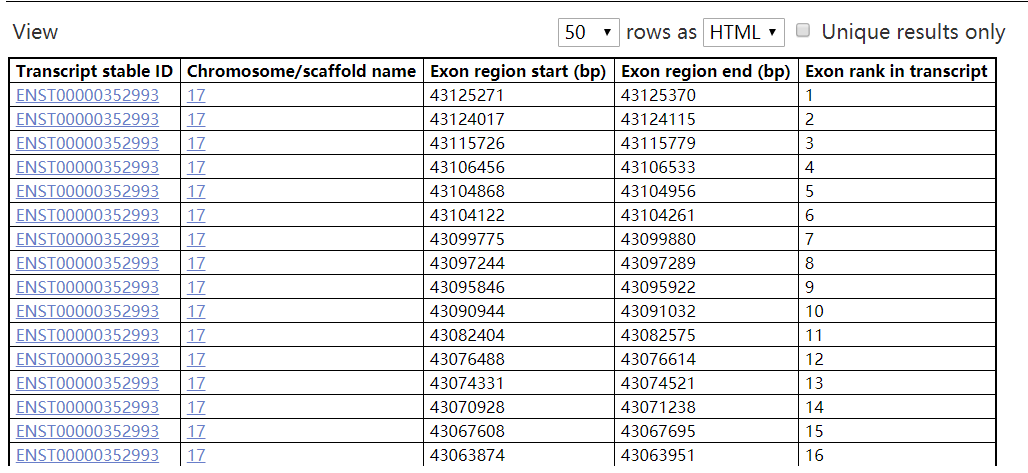
优点很明显,直接得到各个外显子的坐标以及排序,还有R包可以用,貌似python包也有。
三、UCSC
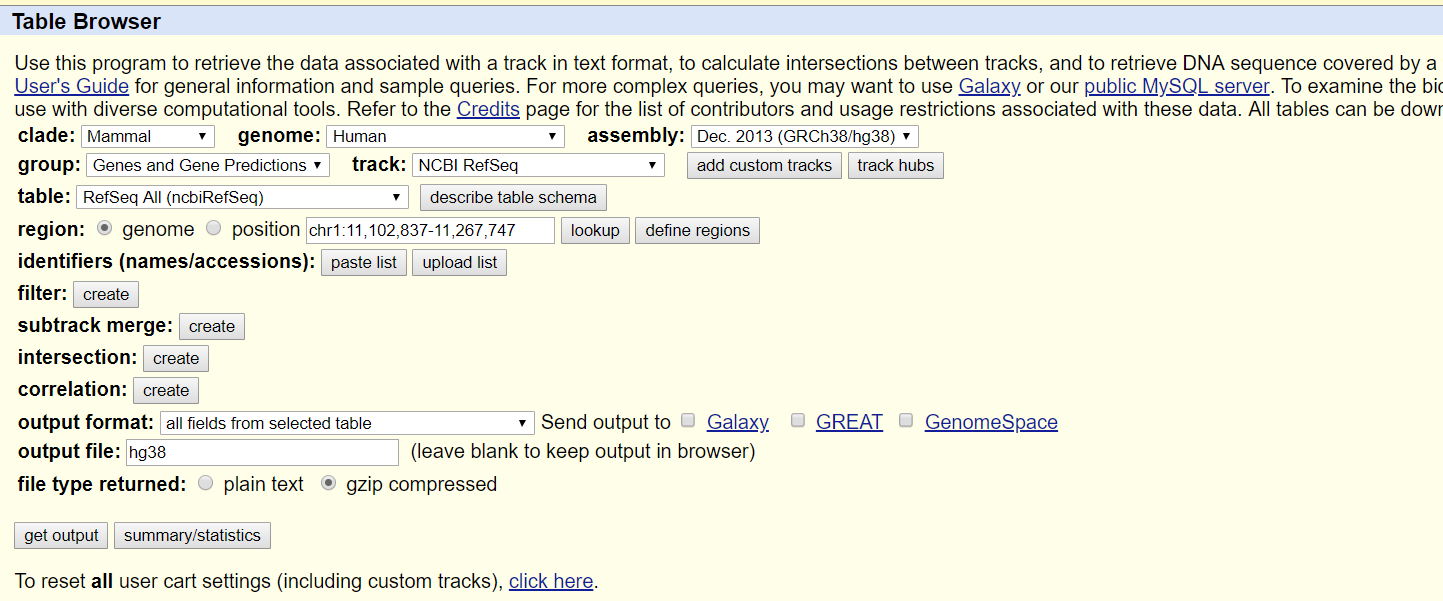
使用UCSC的Table Browser,把数据都下载下来。按照我这样选择。点击get output得到文件。hg38文件大约6m,hg19大约3m。
然后用命令行来查找或者编程查找都可以
zcat hg38.gz | grep BRCA1
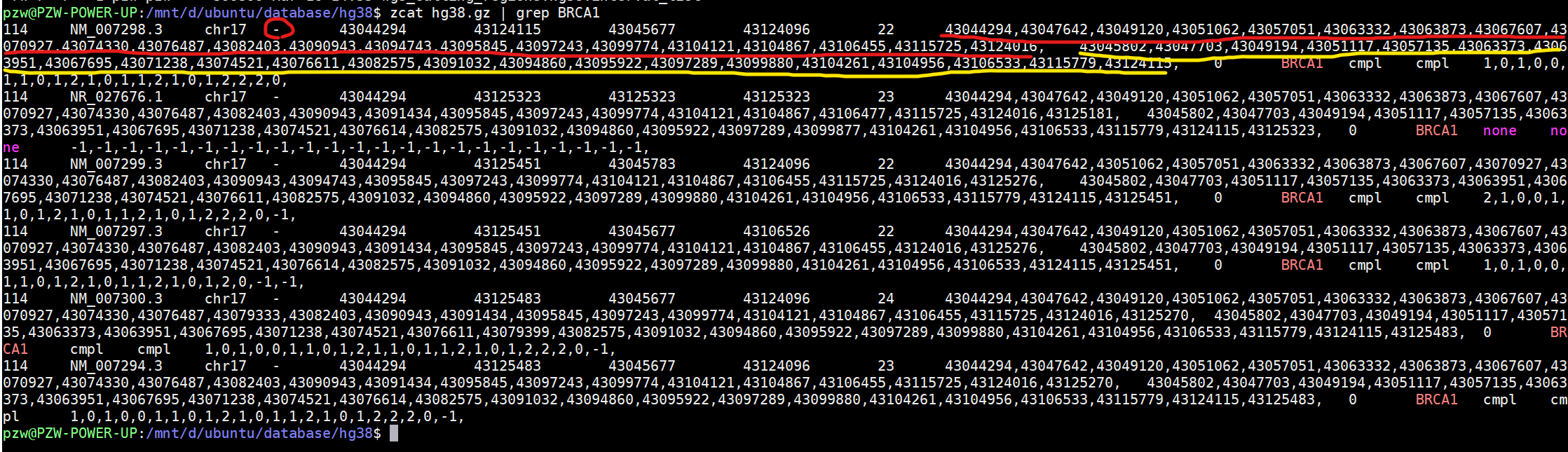
可以看到出来了很多个转录本,以第一个为例,先看到的是“-”表明是反向,红线是每个外显子的start,黄线是每个外显子的end,一一对应。反向所以一号外显子是最后一个咯。
也可以使用cruzdb这个python包,但是这个包不适用于python3。
pip install cruzdb
pip install sqlalchemy
大概应该这样写
from cruzdb import Genome
def GeneExon(gene):
g = Genome(db="hg38")
GENE = g.refGene.filter_by(name2=gene).first()
exonList = GENE.exons
if GENE.strand == "-":
exonList = exonList[::-1]
n = 0
for i in exonList:
n += 1
a = "Exon" + str(n) + ":"
b = str(GENE).split("\t")[0]
c = str(i[0]).split("L")[0] + "\t" + str(i[1]).split("L")[0]
output = a + "\t" + b + "\t" + c
print(output)
GeneExon("BRCA1")