从illumina公布的文档可以看到目前illumina是怎么进行生信分析的,illumina是通过自有平台把自己的软件都搭起来。不过,部分软件已在GitHub开源,所以我们也是可以用这部分的软件进行流程搭建的。
开始之前
illumina的开源软件打包成了dll,在linux里使用需要安装dotnet。可以参照微软官方教程进行安装。这里我选择ubuntu上的dotnet安装教程。
找到了18.04版本ubuntu的dotnet安装命令,照着装就可以了。
首先将 Microsoft 包签名密钥添加到受信任密钥列表。
wget https://packages.microsoft.com/config/ubuntu/18.04/packages-microsoft-prod.deb -O packages-microsoft-prod.deb
sudo dpkg -i packages-microsoft-prod.deb
然后安装SDK即可
sudo apt-get update
sudo apt-get install -y apt-transport-https
sudo apt-get update
sudo apt-get install -y dotnet-sdk-2.1
illumina的dll均可在其开源项目中找到。
路线图
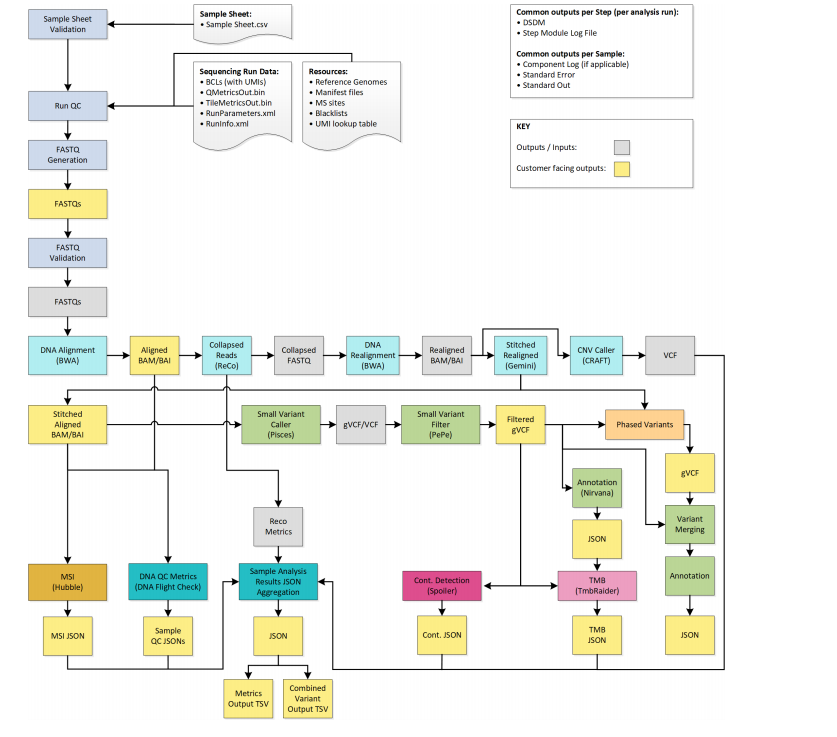
质控
从路线图中看,采取了建立黑名单等方式,在BCL文件中生产fastq文件过程中进行了初步的QC。由于没有找到illumina进行质控的工具,这里使用fastp替代
fastp -i raw_1.fastq.gz -I raw_2.fastq.gz \
-o clean_1.fastq.gz -O clean_2.fastq.gz \
-w 8 -j report.json -h report.html
比对
illumina也是使用bwa进行比对
bwa mem -t 8 hg19.fa clean_1.fastq.gz clean_2.fastq.gz \
-R "@RG\tID:sampleID\tPL:illumina\tSM:tumor\tPU:test" \
| samtools view -bSh - \
| samtools sort -@ 8 - -o 1.bam
samtools index 1.bam
UMI处理
感觉illumina应该是在bcl2fastq的时候就将UMI提取了出来(通过在samplesheet中指定UMI位置),往后使用了Stitcher来合并双端reads的时候顺便形成了UMI的tag,然后后续再去重。由于具体不知道怎么做,因此这里使用fgbio进行替代。 从1.bam文件中获得校正后的2.fastq,然后再次使用bwa比对,获得2.bam。另外,由于上游使用了fastp进行质控,因此这里用gencore进行替代可能更方便
java -jar fgbio.jar FastqToBam \
--input clean_1.fastq.gz clean_2.fastq.gz \
--read-structures 5M5S+T +T \
-o unmapped.bam
java -jar fgbio.jar ExtractUmisFromBam \
-i 1.bam \
-o 2.bam \
-r 5M5S+T +T \
-s RX \
-t ZA ZB
samtools fastq unmapped -1 clean.UMI.1.fastq.gz -2 clean.UMI.2.fastq.gz
bwa mem -t 8 hg19.fa clean.UMI.1.fastq.gz clean.UMI.2.fastq.gz \
-R "@RG\tID:sampleID\tPL:illumina\tSM:tumor\tPU:test" \
| samtools view -bSh - \
| samtools sort -@ 8 - -o umi.bam
java -jar picard.jar MergeBAMAlignment R=hg19.fa \
UNMAPPED_BAM=unmapped.bam \
ALIGNED_BAM=umi.bam \
O=umi.merge.bam \
CREATE_INDEX=true \
MAX_GAPS=-1 \
ALIGNER_PROPER_PAIR_FLAGS=true \
VALIDATION_STRINGENCY=SILENT \
SO=coordinate \
ATTRIBUTES_TO_RETAIN=XS
java -jar fgbio.jar GroupReadsByUmi \
--input=umi.merge.bam \
--output=umi.group.bam \
--strategy=paired --min-map-q=20 --edits=1 --raw-tag=RX
java -jar fgbio.jar CallMolecularConsensusReads \
--min-reads=1 \
--min-input-base-quality=20 \
--input=umi.group.bam \
--output=umi.consensus.bam
samtools fastq umi.consensus.bam | bwa mem -t 8 \
-p hg19.fa /dev/stdin \
| samtools view -bSh - \
| samtools sort -@ 8 - -o consensus.mapped.bam
java -jar picard.jar MergeBAMAlignment \
R=hg19.fa \
UNMAPPED_BAM=umi.consensus.bam \
ALIGNED_BAM=consensus.mapped.bam \
O=consensus.merge.bam \
CREATE_INDEX=true \
MAX_GAPS=-1 \
ALIGNER_PROPER_PAIR_FLAGS=true \
VALIDATION_STRINGENCY=SLIENT \
SO=coordinate \
ATTRIBUTES_TO_RETAIN=XS
java -jar fgbio.jar FilterConsensusReads \
--input=consensus.merge.bam \
--output=consensus.merge.filter.bam \
--ref=hg19.fa --min-reads=2 \
--max-read-error-rate=0.05 \
--max-base-error-rate=0.1 \
--min-base-quality=30 \
--max-no-call-fraction=0.20
java -jar fgbio.jar ClipBAM \
--input=consensus.merge.filter.bam \
--output=consensus.merge.filter.clip.bam \
--ref=hg19.fa --soft-clip=false \
--clip-overlapping-reads=true
indel区域重校正
使用Gemini软件进行重校正,并合并双端reads,再进行过滤后生成3.bam文件。包括后续的步骤,都需要对参考基因组创建一个xml作为索引
创建索引,需将hg19.fa放在hg19/文件夹下
dotnet CreateGenomeSizeFile.dll \
-g hg19/ \
-s "Homo sapiens (UCSC hg19)" \
-o hg19/
使用Gemini
dotnet Gemini.dll --bam 2.bam --genome hg19/ \
--samtools samtools_path/ \
--outFolder output_path/ --numThreads 8
变异检测
使用pisces进行变异检测。此步将会把reads的来源(包括来源自merge后的reads等信息)等注释到生成的1.vcf文件中(实际会在output文件夹下生成与bam文件同名的genome.vcf文件)。随后,使用Psara检测位于内含子/外显子边界的变异,生成2.vcf文件。
dotnet Pisces.dll -b 3.bam \
-g hg19/ -o output/ -t 8 \
-i target.bed --mindp 10000 --minvf 20
dotnet Psara.dll --vcf 1.vcf --roi target.bed -o output/
变异过滤
使用Pepe进行过滤。未找到此软件,所以这里使用bcftools进行替代,可根据实际自行设定参数
bcftools view -i "FILTER='PASS'" 2.vcf > 3.vcf
CNV检测
使用CRAFT对重校正后的2.bam文件进行CNV检测。CRAFT会统计每个靶区域的counts分布,进行标准化并计算log2FC,并获得每个靶区域的CNV状态。标准化校正中会考虑测序深度、靶区域大小、PCR重复、探针效率、GC比例以及DNA类型等。同时,需要使用正常的FFPE和cfDNA样本集建立基线。最终获得cnv.vcf文件。
未找到CRAFT软件。可使用cnvkit代替
cnvkit.py batch tumor.bam \
--normal normal.bam \
--method hybrid \
--target refs.bed \
--annotate refFlat.txt \
--output-reference reference.cnn \
--output-dir output/ \
--disgram --scatter \
-p 8
MNV检测
使用Scylla进行多核苷酸突变检测。使用此软件主要是为了检测EGFR 19号外显子缺失。Scylla将使用重校正后的2.bam文件以及Psara获得的2.vcf文件作为输入,校正特定的区域,并生成校正后的mnv.vcf文件
dotnet Scylla.dll --bam 2.bam --vcf 2.vcf -t 8 -o output/ -g hg19/
合并Vcf
流程最终会将3.vcf以及mnv.vcf进行合并。拥有相同的chromosome、position、reference allele、alternative allele的重复位点会被删除。在mnv.vcf中,只有下列表格中的EGFR突变会被保留,其他EGFR突变均会被过滤
| chromosome | Position | Reference Allele | Alternative Allele |
|---|---|---|---|
| chr7 | 55242463 | AAGGAATTAAGAGAAG | A |
| chr7 | 55242464 | AGGAATTAAGAGA | A |
| chr7 | 55242464 | AGGAATTAAGAGAAGC | A |
| chr7 | 55242465 | GGAATTAAGAGAAGCA | G |
| chr7 | 55242467 | AATTAAGAGAAGCAAC | A |
| chr7 | 55242469 | TTAAGAGAAGCAACATCTC | T |
| chr7 | 55242468 | ATTAAGAGAAGCAACATCT | A |
| chr7 | 55242466 | GAATTAAGAGAAGCAA | G |
| chr7 | 55242465 | GGAATTAAGA | G |
| chr7 | 55242469 | TTAAGAGAAGCAA | T |
| chr7 | 55242462 | CAAGGAATTAAGAGAA | C |
| chr7 | 55242466 | GAATTAAGAGAAGCAACAT | G |
| chr7 | 55242482 | CATCTCCGAAAGCCAACAAGGAAAT | C |
| chr7 | 55242465 | GGAATTAAGAGAAGCAACA | G |
| chr7 | 55242467 | AATTAAGAGAAGCAACATC | A |
| chr7 | 55242469 | TTAAGAGAAG | C |
| chr7 | 55242467 | AATTAAGAGAAGCAACATC | T |
| chr7 | 55242469 | TTAAGAGAAGCAA | C |
| chr7 | 55242467 | TTAAGAGAAGCAA | TTGCT |
| chr7 | 55242468 | ATTAAGAGAAG | GC |
| chr7 | 55242469 | TTAAGAGAAGCAACATCTCC | CA |
| chr7 | 55242465 | GGAATTAAGAGAAG | AATTC |
| chr7 | 55242465 | GGAATTAAGAGAAGCAAC | AAT |
| chr7 | 55242467 | AATTAAGAGAAGCAAC | T |
| chr7 | 55242467 | AATTAAGAGAAGCAACATCTC | TCT |
| chr7 | 55242469 | TTAAGAGAAGCAACATCT | CAA |
| chr7 | 55242465 | GGAATTAAGAGAAGCAA | AATTC |
| chr7 | 55242465 | GGAATTAAGAGAAGCAACATC | AAT |
| chr7 | 55242468 | ATTAAGAGAAGCAAC | GCA |
注释
illumina使用Nirvana进行注释,输入文件为3.vcf文件,输出文件为1.json文件。
需要使用Nirvana自行下载注释数据库。
dotnet Nirvana.dll \
-i 3.vcf \
-r GRCh37.dat \
--sd SupplementaryAnnotation \
--cache /Cache/GRCh37/Both \
-o /output/4
其他
TMB计算可通过自建统计脚本进行
\[TMB = 合格变异数 / Panel大小 (单位: muts/Mb)\]msi分析可使用msisensor2等软件进行
msisensor2 msi -M models_hg19 -t 3.bam -o msi/
融合检测
illumina使用Manta进行融合检测,生成vcf文件
python configManta.py \
--tumorBam tumor.bam \
--referenceFasta hg19.fa \
--exome \
--generateEvidenceBam \
--runDir output/
python runWorkflow.py