数据获取
TCGA的MC3数据集包含了33个癌种的体细胞突变,在分析TMB时,可以使用MC3数据集的数据进行验证。
在这个公开页面中下载MC3的数据,没有权限只能下载Open-Access的mc3.v0.2.8.PUBLIC.maf.gz。同时下载bed文件,下载样本信息文件。
初步整理
下载下来的数据解压后是maf文件,可以使用maftools进行分析,为了适用于自己的流程,这里使用maf2vcf将其转换回vcf格式。(这里maf2vcf是会直接将maf文件拆分回多个vcf文件的,但是我感觉有点慢,因此自行先将maf根据样本名拆分成了多个小maf,再使用maf2vcf来转换到vcf了,这样方便使用多个线程)。
perl maf2vcf.pl --input-maf mc3.v0.2.8.PUBLIC.maf --output-dir outputDir --ref-fasta GRCh37.fa
注意,结果vcf中,是包含非PASS的突变的,要进行过滤,最终保留至少包含1个PASS突变的样本。
bcftools view -f 'PASS' input.vcf > output.vcf
下载下来的bed文件,需要使用cds的bed取一次交集,才是外显子bed
bedtools intersect -a gencode.v19.basic.exome.bed -b cds.bed | bedtools sort -i - | bedtools merge -i - > exome.bed
然后对vcf文件取交集,同理,这步可以使用自己的target bed来取,获得vcf后,就可以回归到大部分人的流程中,使用snpeff、annovar、vep等软件进行注释了,注释好后按照自身设定的条件进行过滤即可。
bedtools intersect -a input.vcf -b exome.bed -header > output.vcf
TMB计算
过滤条件和分母大小(按照NCCL的预研,分母大小除了仅统计外显子区外,还需要删除部分阴性区域及弱阳区域,这里先不管)都参考这篇文章。
以上方式获得了MC3数据在WES中的TMB值,之后以自己的Target再获得一个Panel中的TMB值,将数据随机分为两组,其中一组用于Panel TMB及WES TMB的拟合,另外一组用于计算测试。
我整理好的表格,格式如下
| SampleID | TumorType | WES | Target |
|---|---|---|---|
| TCGA-02-0003 | GBM | 0.69 | 0.98 |
| TCGA-02-0033 | GBM | 0.47 | 1.95 |
| TCGA-02-0047 | GBM | 1.31 | 2.93 |
线性回归
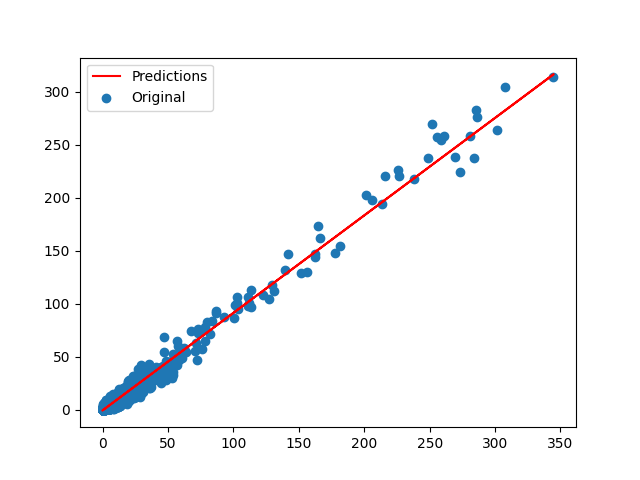
其实用excel就可以做,但这里是用python statsmodels做的,获得Target TMB与WES TMB的回归方程。
import pandas as pd
import matplotlib.pyplot as plt
import statsmodels.api as sm
# 读数据
df = pd.read_excel("TCGA_MC3.xlsx", sheet_name="Train", header=0)
y = df["WES"]
x = df["Target"]
# 构筑模型
x = sm.add_constant(x)
model = sm.OLS(y, x).fit()
summary = model.summary()
print(summary)
summary_df = pd.read_html(summary.tables[1].as_html(), header=0, index_col=0)[0]
b = summary_df.iat[0, 0]
k = summary_df.iat[1, 0]
if b < 0:
print("functions: y = " + str(k) + "x - " + str(abs(b)))
else:
print("functions: y = " + str(k) + "x + " + str(b))
# 画图
predicts = model.predict()
x = df["Target"]
plt.scatter(x, y, label="Original")
plt.plot(x, predicts, color = "red", label='Predictions')
plt.legend()
plt.show()
单个癌种
一般采用四分位数计算阈值,即将TMB值从高往低排,取排在25%的值作为TMB-H的阈值,采用二分类,低于阈值的为TMB-L(不知道要不要先把0值去掉)。
这里以肺腺癌的数据为例。
import pandas as pd
df = pd.read_excel("TCGA_MC3.xlsx", sheet_name="Train", header=0)
df_select = df[df["TumorType"] == "LUAD"]
df_sort = df_select.sort_values(by=["WES"], ascending=False)
sampleAmount = len(df_sort)
top_25_percent = int(sampleAmount / 4)
cutoff_index = df_sort.index[top_25_percent - 1]
cutoff = df_sort.at[cutoff_index, "WES"]
print(cutoff)
df_sort["Judge"] = "TMB-L"
df_sort["Judge"] = df_sort["Judge"].where(df["WES"] < cutoff, "TMB-H")
df_sort.to_excel("TCGA.LUAD.xlsx", sheet_name="LUAD", header=True, index=False)
拿这个阈值得到的TMB结果去分别对测试组的WES结果(作为实际值)和经回归公式校正后的Target结果进行比较,得到准确率结果。
箱线图
用ggplot2画箱线图及小提琴图
library(openxlsx)
library(ggplot2)
###########################
cancerType = "LUAD"
TMB <- 8.2
###########################
ms <- read.xlsx(xlsxFile="TCGA_MC3.xlsx", sheet=3, colNames=TRUE)
selectType = ms[ms$TumorType == cancerType, ]
# selectType = selectType[selectType$WES 3, ]
# selectType = selectType[selectType$WES < 20, ]
# plot
e1 <- ggplot(selectType, aes(x=TumorType, y=WES
e2 <- e1 + geom_violin(fill="cornflowerblue", color="cornflowerblue", scale="width") + coord_flip()
e3 <- e2 + stat_boxplot(geom="errorbar", width=0.15, aes())
e4 <- e3 + geom_boxplot(width=0.2, outlier.colour="cornflowerblue", fill="cornflowerblue")
e5 <- e4 + geom_hline(aes(yintercept=TMB), colour="#990000", linetype="dashed", size=1)
e6 <- e5 + theme(panel.border=element_blank()) + ylab("Tumor Mutation Burden") + xlab("")
e6 + theme_minimal()
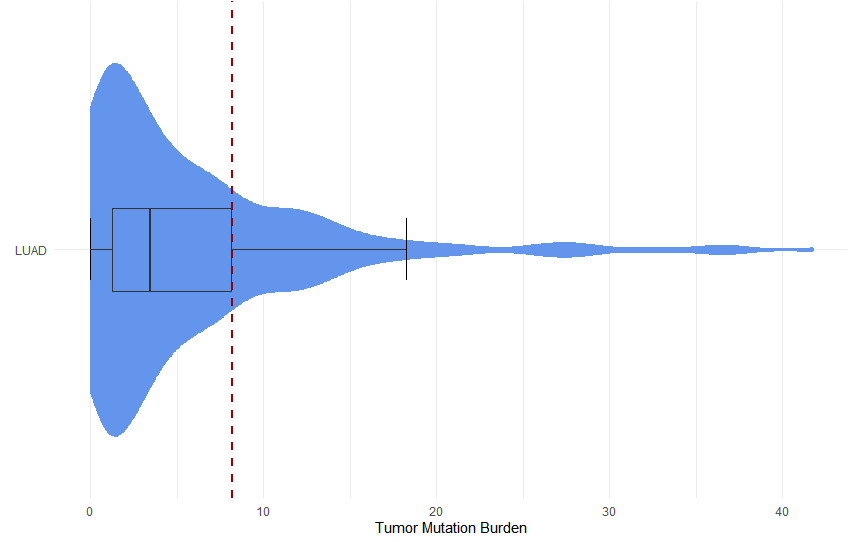
效果不是很好,所以上游可能还需要过滤接近0的值和一些离散值。