Call SNP/Indel找变异
找变异基本上是重中之重了。有GATK4 best practice提供的复杂的方法,也有使用属于samtools全家桶的bcftools找变异的方法。这里将会介绍这两种方法。最终结果都是得到vcf文件。samtools这里有一篇介绍vcf格式的pdf。
使用GATK4进行call snp/indel
虽说是call snp,不过当然,也会call indel。
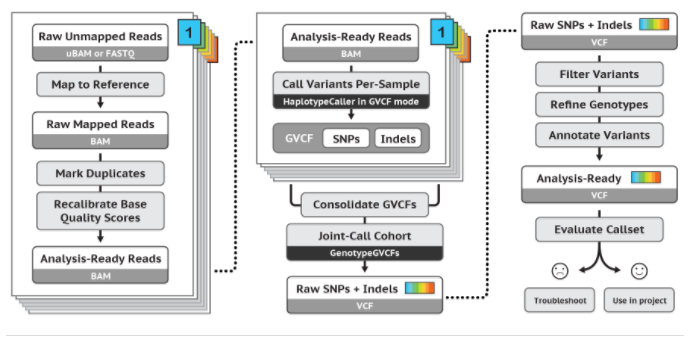
1 2 3 4 | |
另外,当输入是多样本时,其实gatk可以先输出GVCF文件,再将它们合并。
使用bcftools进行call snp/indel
bcftools的流程已经有一篇不错的教程,非常值得参考。bcf其实就是vcf的二进制格式,这里仍然输出我们更加熟悉的vcf。 bcftools mpileup会产生超级大的中间文件,所以这里建议用管道往后进行call snp。另外,Varscan这个工具也可以使用。
1 2 3 4 | |
使用freebayes进行call snp/indel
freebayes也是一款不错的软件。
1 | |