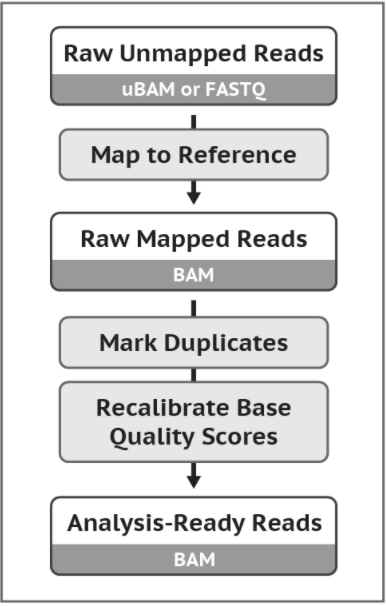
GATK4的推荐流程哦。这个流程是从ubam文件或fastq文件得到可以用于找变异的bam文件。 以后GATK大概会把bwa和picard完全内嵌。 下面的命令其实还有很多参数的,不过一般来说用这些参数就够。 原始数据要用经过了质控之后的数据!
放两个用作测试学习的原始数据。 点这里下载!
#1 先把需要用到的软件还有数据库下载好 首先是软件。当然是要用最新的啦,跟上时代。
然后是数据库。 用的是GATK提供的hg19。 地址在这:点击进入
#2 把软件都加到环境中,或者在shell脚本中设定好 像这样。
reference=/database/hg19/ucsc.hg19.fasta
indel1=/database/hg19/1000G_phase1.indels.hg19.vcf
indel2=/database/hg19/Mills_and_1000G_gold_standard.indels.hg19.vcf
gatk=/software/gatk-4.0.0.0/gatk
bwa=/software/bwa0.7.17/bwa
picard=/software/picard2.17.4/picard.jar
sample1=/project/test/data/test-1.fastq.gz
sample2=/project/test/data/test-2.fastq.gz
第一次用软件,还要建立好索引文件。以后不需要。
$bwa index $reference
java -jar $picard CreateSequenceDictionary \
R=$reference
#3 比对到参考序列
bwa mem -t 6 $reference $sample1 $sample2 > sample.sam
# -t 6 表示用6个线程来跑
#4 对比对结果进行排序 按croodinate来排,就是根据染色体号还有位置排。
java -jar $picard SortSam \
I=sample.sam \
O=sample.sorted.bam \
SO=coordinate
#5 去掉重复序列
java -jar $picard MarkDuplicates \
I=sample.sorted.bam \
O=sample.marked_dup.bam \
M=marked_dup.txt
#6 加上read group信息 不加的话后面GATK会报错的。
java -jar $picard AddOrReplaceReadGroups \
I=sample.marked_dup.bam \
O=sample.addhead.bam \
RGID=4 \
RGLB=lib1 \
RGPL=illumina \
RGPU=unit1 \
RGSM=20
# 这里后面几个RG信息都按自己的实际情况填哦
#7 对自己的bam建立索引
java -jar $picard BuildBamIndex \
I=sample.addhead.bam
#8 重新比对indel区域 这里GATK4和以前的版本不一样了,运用了machine learning。
$gatk BaseRecalibrator \
-I sample.addhead.bam \
-R $reference \
--known-sites $indel1 \
--known-sites $indel2 \
-O recal_data.table
#9 校正质量值
$gatk ApplyBQSR \
-R $reference \
-I sample.addhead.bam \
--bqsr-recal-file recal_data.table \
-O sample.pre.analysis.bam
这样,就得到了一个可以供后续使用的bam文件。