这篇是Hisat2+FeatureCounts+DESeq2的流程。
更详细的流程请点击这里。
featureCounts是一个用来统计count数的软件,运行的速度飞快,比之前用的htseq-count快了好多好多。 照例先说一下怎么下载这个软件:
wget https://jaist.dl.sourceforge.net/project/subread/subread-1.6.2/subread-1.6.2-Linux-x86_64.tar.gz
tar -zxvf subread-1.6.2-Linux-x86_64.tar.gz
cd subread-1.6.2-Linux-x86_64/bin
./featureCounts -h
然后来说这次的流程。 照旧用Hisat2来比对出Bam文件之后。 使用featureCounts统计:
featureCounts \
-T 16 \
-p \
-t exon \
-g gene_id \
-a Homo_sapiens.GRCh38.92.chr_patch_hapl_scaff.gtf \
-o all_feature.txt \
1.sort.bam \
2.sort.bam \
3.sort.bam \
4.sort.bam
# -T 使用的线程数
# -p 如果是paird end 就用
# -t 将exon作为一个feature
# -g 将gene_id作为一个feature
# -a 参考的gtf/gff
# -o 输出文件
# 最后加上bam文件,有几个就加几个
然后会得到两个文件,一个是结果,一个是结果的summary。 接下来就可以用DESeq2对结果进行愉快的操作了。 使用R。 我这次的样本有6个。
library(DESeq2)
## 数据预处理
sampleNames <- c("10A_1", "10A_2", "10A_3", "7_1", "7_2", "7_3")
# 第一行是命令信息,所以跳过
data <- read.table("all_feature.txt", header=TRUE, quote="\t", skip=1)
# 前六列分别是Geneid Chr Start End Strand Length
# 我们要的是count数,所以从第七列开始
names(data)[7:12] <- sampleNames
countData <- as.matrix(data[7:12])
rownames(countData) <- data$Geneid
database <- data.frame(name=sampleNames, condition=c("10A", "10A", "10A", "7", "7", "7"))
rownames(database) <- sampleNames
## 设置分组信息并构建dds对象
dds <- DESeqDataSetFromMatrix(countData, colData=database, design= ~ condition)
dds <- dds[ rowSums(counts(dds)) > 1, ]
## 使用DESeq函数估计离散度,然后差异分析获得res对象
dds <- DESeq(dds)
res <- results(dds)
write.csv(res, "res_des_output.csv")
resdata <- merge(as.data.frame(res), as.data.frame(counts(dds, normalized=TRUE)),by="row.names",sort=FALSE)
write.csv(resdata, "all_des_output.csv", row.names=FALSE)
输出两个文件,一个只有差异统计的结果,一个包含各个样本的结果。 这样就完成了DESeq2了。
接下来是作图: 一、MA图 MA图是拿来展示数据表达是否异常,现在一般都不用了。
# library(DESeq2)
plotMA(res, main="DESeq2", ylim=c(-2, 2))
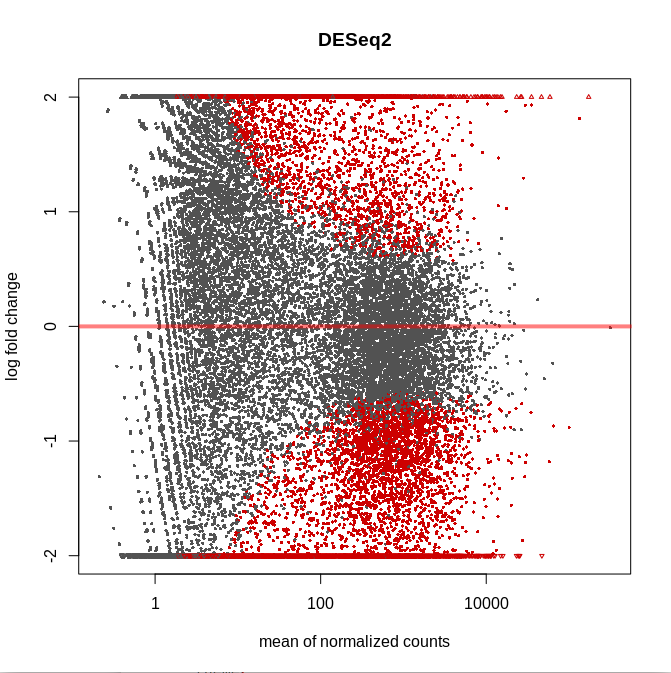
二、火山图 可以非常直观且合理地筛选出在两样本间发生差异表达的基因。
library(ggplot2)
# 这里的resdata也可以用res_des_output.csv这个结果重新导入哦。
# 现在就是用的前面做DESeq的时候的resdata。
resdata$change <- as.factor(
ifelse(
resdata$padj<0.01 & abs(resdata$log2FoldChange)>1,
ifelse(resdata$log2FoldChange>1, "Up", "Down"),
"NoDiff"
)
)
valcano <- ggplot(data=resdata, aes(x=log2FoldChange, y=-log10(padj), color=change)) +
geom_point(alpha=0.8, size=1) +
theme_bw(base_size=15) +
theme(
panel.grid.minor=element_blank(),
panel.grid.major=element_blank()
) +
ggtitle("DESeq2 Valcano") +
scale_color_manual(name="", values=c("red", "green", "black"), limits=c("Up", "Down", "NoDiff")) +
geom_vline(xintercept=c(-1, 1), lty=2, col="gray", lwd=0.5) +
geom_hline(yintercept=-log10(0.01), lty=2, col="gray", lwd=0.5)
valcano
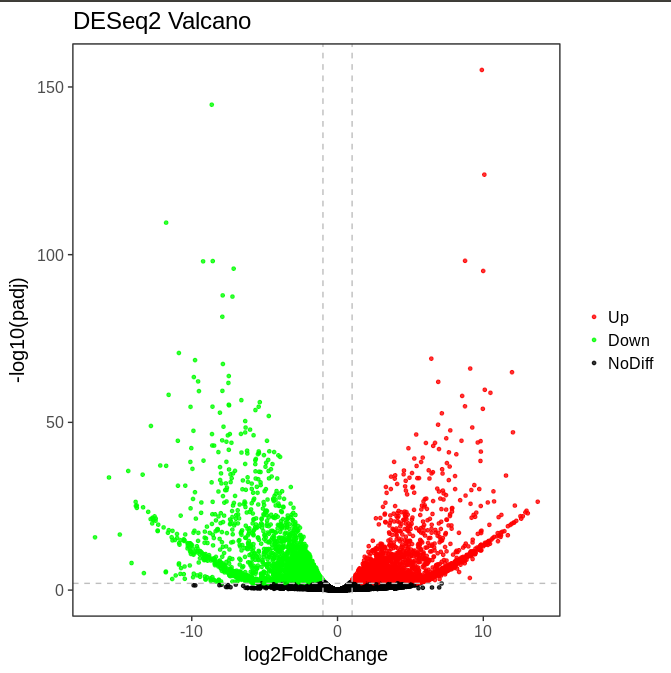
三、PCA图 就是主成分分析。是把数据降维之后进行分析。PC1和PC2分别是贡献率第一的主成分和贡献率第二的主成分。
# library(ggplot2)
rld <- rlog(dds)
pcaData <- plotPCA(rld, intgroup=c("condition", "name"), returnData=T)
percentVar <- round(100*attr(pcaData, "percentVar"))
pca <- ggplot(pcaData, aes(PC1, PC2, color=condition, shape=name)) +
geom_point(size=3) +
ggtitle("DESeq2 PCA") +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance"))
pca
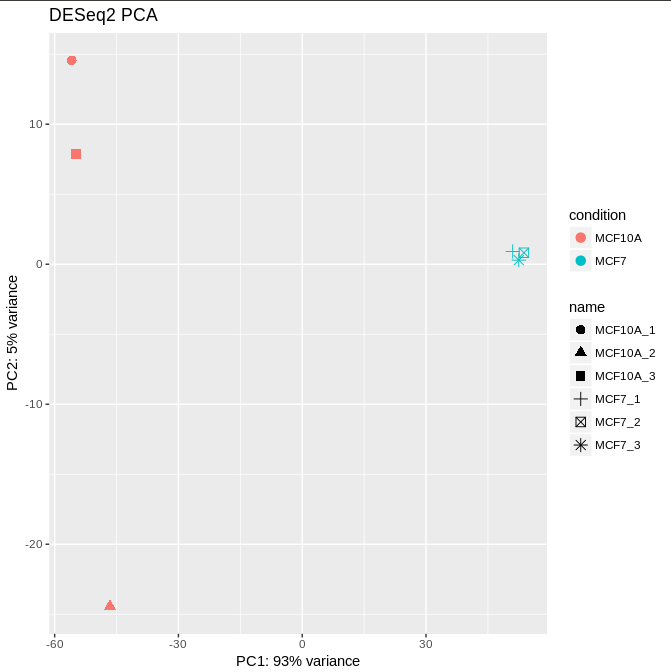
四、热图 heatmap 实现这基因表达模式可视化的需求。 从这里可以看到这6个样本的表达差异。
library(pheatmap)
select <- order(rowMeans(counts(dds, normalized=T)), decreasing=T)[1:1000]
nt <- normTransform(dds)
log2.norm.counts <- assay(nt)[select,]
df <- as.data.frame(colData(dds)[, c("name", "condition")])
pheatmap(log2.norm.counts, cluster_rows=T, show_rownames=F, cluster_cols=T, annotation_col=df, fontsize=6)
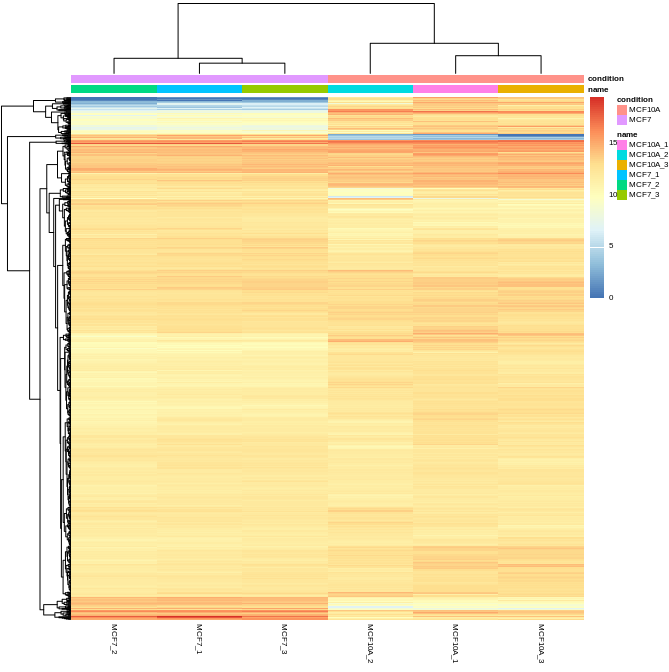
RNA-seq到这里暂告一段落啦!