HLA-HD是一个基于NGS数据的HLA分型软件。除了对常规的HLA-A、-B、-C进行分型后,还可以对-G、-E这些基因进行分型。
可以到官网中留下邮件信息,申请下载。
在下载页面,会有软件的安装与其他介绍,在下载后,首先要确保自身环境变量中有bowtie2,因为软件是使用bowtie2进行比对的。
安装较为简单,下载解压后,进入到软件目录中
sh install.sh
安装后,需要将bin文件夹目录加入到环境变量中。
接下来需要更新数据库,软件自带的数据库是3.15.0,但目前HLA数据库已更新到3.41.0。差异较大。 软件提供了一个更新的方法。
sh update.dictionary.sh
但因为国内网络原因,下载需要的hla.dat文件时,速度较慢。这时可以把updata.dictionary.sh文件中的wget命令注释掉,然后使用下面命令下载hla.dat,该文件需要放置于软件根目录中。接着再运行更新程序。
wget https://media.githubusercontent.com/media/ANHIG/IMGTHLA/Latest/hla.dat
sh update.dictionary.sh
在运行之前,需要检查系统可同时打开的文件数
ulimit -Sa
如果数值小于1024,需要调整到1024
ulimit -n 1024
分析时,需求的输入文件是fastq格式,不能使用压缩文件,因此,需要预先对fastq.gz文件进行解压。
运行命令
hlahd.sh -t 2 -m 100 -c 0.95 -f freq_data fastq1 fastq2 \
HLA_gene.split.txt dictionary sampleID output_dir
其中,-t线程数,-m最短reads长度,-c过滤比例。如果需要分析HLA-DPA2、-T、-W、-Y等分型,需要修改HLA_gene.split.3.32.0.txt文件。输出文件夹需要自行创建。
我用了HLA reporter的数据作为测试数据。
wget http://paed.hku.hk/genome/software/HLAgm_wes.zip
下载下来是bam文件,使用samtools按名称排序再使用bedtools还原为fastq文件就可以了。
samtools sort -n -@ 8 sample.bam
bedtools bamtofastq -i sample.bam -fq sample.R1.fastq -fq2 sample.R2.fastq
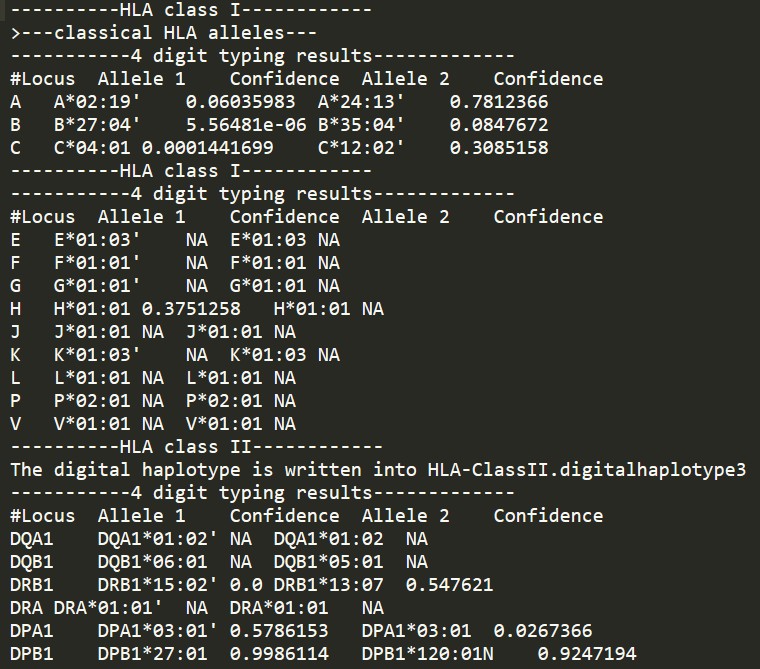
最终在输出文件夹下的result文件夹中,会有一个sampleID_final.result.txt文件,展示所有结果。
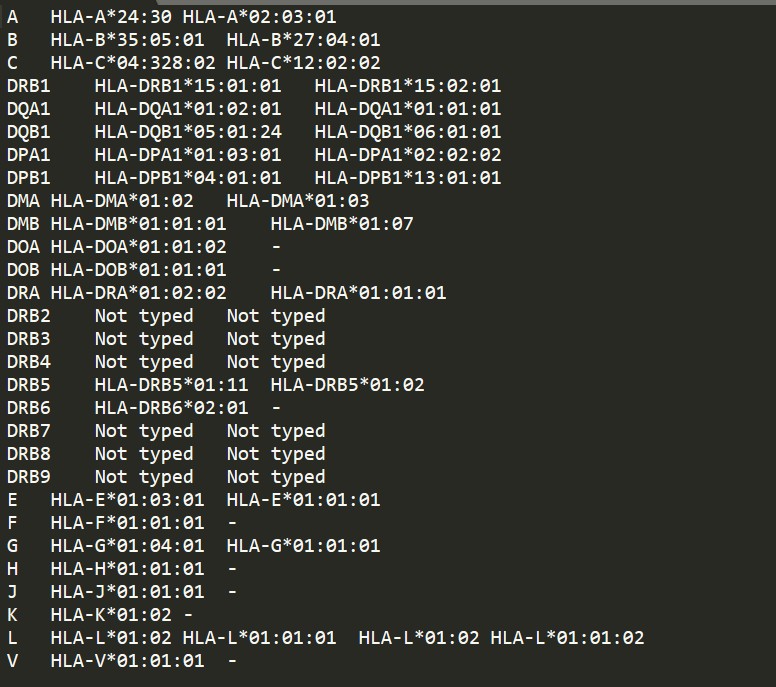
结果中看,“-”应该是无法分型,而Not typed应该是没有进行分型。但是有些分型有4个结果,不太理解。
这个样本的HLA-B检出了高强直风险的HLA-B*27:04。
和seq2HLA的结果对照一下,还是有不少差异呢,接下来需要评估一下哪个比较准咯。