基于SNP的祖源分析
祖源分析
参考23andme,使用47个人群数据进行祖源分析分类。这47个人群合计有超过14812个样本,通过进行主成分分析,可以将单个样本的结果分类到这47个人群之中(所以只是最相似)。显然,检测公司可以获得更多的非公开数据优化他们的模型,而我们只能使用公开数据了。
admixture应该是被用得最多的基于SNP进行祖源分析的软件。
这里使用admix进行分析,这个软件预设了不同的模型,使用admixture算法对23andme格式的输入文件进行祖源分析。提供的模型最大的人群数目也是47个。
admix -f input.txt -v 23andme -m K47
我感觉结果还不错的。
父系单倍群
单倍群是由一组特定的遗传标记(SNP)定义的群体,这些标记在进化过程中稳定遗传。Y单倍群反映了人类父系祖先的分支关系。Y单倍群的共同祖先被称为“Y染色体亚当”,是所有现代男性Y染色体的最近共同祖先,大约生活在20万至30万年前的非洲。
| 常见的Y单倍群 | 描述 |
|---|---|
| R1b | 西欧最常见的单倍群,与凯尔特人和日耳曼人相关 |
| R1a | 东欧和南亚常见的单倍群,与斯拉夫人和印欧语系相关 |
| O | 东亚最常见的单倍群,与汉人、日本人、韩国人相关 |
| E1b1b | 北非和东非常见的单倍群,与柏柏尔人和阿拉伯人相关 |
| Q | 美洲原住民和西伯利亚族群的主要单倍群 |
ISOGG上有大量Y染色体的相关工具,不知道哪个工具好用,只能从发布和更新时间入手,找新的工具进行测试。
首先下载最新的SNP索引表(截止到文章日期,版本15.73,发布时间是20200711),有两个版本,分别是中国特供版和普通版,说是中国版本对中国单倍群进行了更细的分类。
我使用haploGrouper进行分型,输入文件是vcf格式,数据库版本是2019的,应该也够用。
python3 haploGrouper.py \
-v data/test.vcf \
-t data/chrY_isogg2019_tree.txt \
-l data/chrY_isogg2019-decode1_loci_b37.txt \
-o docs/test.Y.txt
母系单倍群
母系的单倍群和Y类似,但使用的是线粒体(MT)上的位点进行,因为理论上线粒体的遗传都来自于母亲。MT单倍群的共同祖先被称为“线粒体夏娃”,大约生活在20万年前的非洲。phylotree上公布了MT的发生树,目前的最新版本是17.2。
注意,线粒体的参考基因组是rCRS版本,即16569bp长度的那个。
使用haploGrouper进行分析,数据库版本是17,应该是最新的。
python3 haploGrouper.py \
-v data/test.vcf \
-t data/chrMT_phylotree17_tree.txt \
-l data/chrMT_phylotree17_loci.txt \
-o docs/test.MT.txt
尼安德特比例
研究认为尼安德特人是智人的一个分支,这个方案是计算基因组中包含的尼安德特人基因比例(一般是1%~4%,非洲人除外,因为研究认为现代智人在走出非洲后才与尼安德特人合流)。
分析的逻辑应该是找到尼安德特人与现代人相比,特有的突变。然后分析样本的基因组里包含这些突变的比例。所以,首先还是要选择一个参考的尼人群体。这里选择Vindija33.19这个样本,是因为研究说这个样本更接近与现代人杂交的尼人群体。
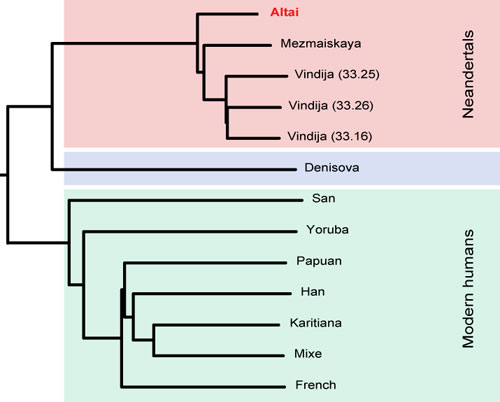
注意,除了计算比例外,23andme还有另外一个逻辑。23andme是预设了2855个(2872个?)已验证来源于尼人的突变,然后最后返回一个个人样本中检出的尼人突变数目的结果。具体查看SNPedia此篇。
应该下载所有染色体的vcf进行分析。我目前想到的方案是,下载了尼人vcf后,去掉所有野生型位点,然后把剩余的位点拿去注释gnomAD,去掉所有的有人群频率注释的位点,然后这时剩余的位点作为尼人特有突变。
PMC4072735高引文章也给了一些位点,但是文章上的链接已经打不开了,不过我找到了最新的网址。
VCF
我写了一个转换23andme格式数据到vcf格式的python脚本,使用方法如下
python3 convert_23andme_to_vcf.py -i 23andme.txt -o 23andme.vcf -r b37.fasta