DADA2是一个通过错误率模型,衡量扩增子序列是否来自模板的算法。 和通过查找样品中物种的组成,比较OTU数据库的聚类算法不同,DADA2采取的是降噪算法。 然后作者还发了文章说明降噪算法比聚类算法要好。PMID 28731476
基于技术向前走的原因,先不去研究OTU方法了。直接来DADA2。 DADA2是一个R包,直接在R中安装。
source("https://bioconductor.org/biocLite.R")
biocLite("dada2")
下载一批测试数据,按照教程来,下载mothur的测试数据。 我把数据解压进了Rawdata文件夹。 然后开始使用dada2进行分析。
一、读入文件
library(dada2)
# 指定文件夹位置
path <- "~/workspace/AMP/Rawdata"
list.files(path)
# 利用文件的固定命名方式,读取需要的文件名(双端)
fnFs <- sort(list.files(path, pattern="_R1_001.fastq", full.names=TRUE))
fnRs <- sort(list.files(path, pattern="_R2_001.fastq", full.names=TRUE))
# 取得文件名,由于正向和方向名字是一样的,所以对其中一个方向取一次名就可以了
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)
# 查看数据质量
plotQualityProfile(fnFs[1]) # 这里指查看正向的第一个样本
plotQualityProfile(fnRs[1:3]) # 这里指查看反向的第一到三个样本
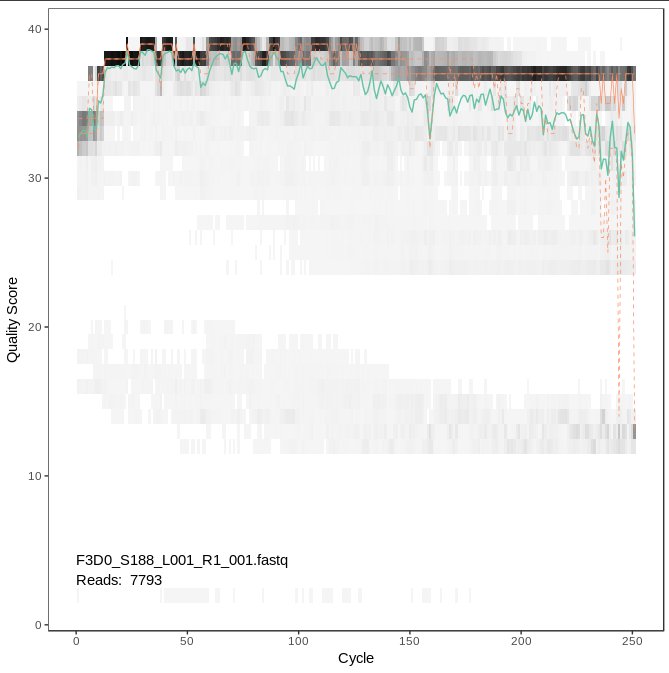 一般的,我们需要的数据也是要求质量在Q20以上,所以,这里需要把后面小于Q20的过滤掉。
一般的,我们需要的数据也是要求质量在Q20以上,所以,这里需要把后面小于Q20的过滤掉。
二、数据过滤
# 指定过滤文件输出的文件夹
filtFs <- file.path("~/workspace/AMP/Cleandata", paste0(sample.names, "_F_filt.fastq.gz"))
filtRs <- file.path("~/workspace/AMP/Cleandata", paste0(sample.names, "_R_filt.fastq.gz"))
# 过滤
out <- filterAndTrim(
fnFs, filtFs, fnRs, filtRs, truncLen=c(240, 160),
maxN=0, maxEE=c(2, 2), truncQ=2, rm.phix=TRUE,
compress=TRUE, multithread=TRUE # 在windows下,multithread设置成FALSE
)
head(out)
一个提示 在过滤后,数据仍然需要有overlap的部分,方便合并! 教程用的是V4数据。 指的是16S rDNA的结构,从5’到3’,分别有10个功能区,就是V1一直到V10。 由于V4-V5的特异性较高,所以一般都是测V4区域。 如果用的是overlap比较低的数据,比如V1-V2或者V3-V4, truncLen的范围就需要更大。 除了truncLen值,maxEE值也需要灵活调整。maxEE为错误容许。如果发现通过的reads数太少,可以调整maxEE。 maxEE和truncLen中,前为正向参数,后为反向参数。 ion torrent数据在filterAndTrim中可加入参数trimLeft=15。
三、计算错误率
前面说了,dada2使用的是一个叫错误率模型的比对方式。
# 分别计算正向和反向
errF <- learnErrors(filtFs, multithread=TRUE)
errR <- learnErrors(filtRs, multithread=TRUE)
# 画出错误率统计图
plotErrors(errF, nominalQ=TRUE)
plotErrors(errR, nominalQ=TRUE)
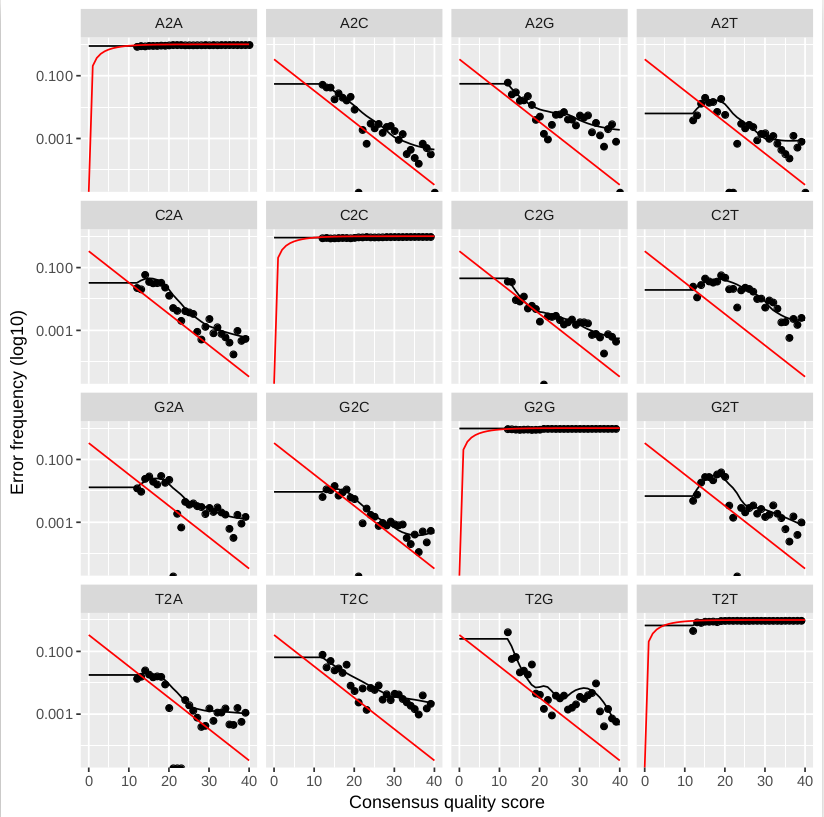 这里表示每个碱基变化方向的错误率,一个黑点就是一个样本。黑线为错误率的收敛线,红线为期望中的错误率。
这里表示每个碱基变化方向的错误率,一个黑点就是一个样本。黑线为错误率的收敛线,红线为期望中的错误率。
四、消除误差
去重复是将所有相同的测序读数组合成“独特序列”,其相应的“丰度”等于具有该独特序列的读数的数量。
# 下面其实是一个批量的操作,如果是处理大文件,内存可能不足,更好的做法是一个一个样本的进行
derepFs <- derepFastq(filtFs, verbose=TRUE)
derepRs <- derepFastq(filtRs, verbose=TRUE)
# 用sample.names来改名
names(derepFs) <- sample.names
names(derepRs) <- sample.names
五、DADA2核心算法
可以理解成根据之前算处理的错误率还有消除误差之后的结果,通过dada算法进行调整。
dadaFs <- dada(derepFs, err=errF, multithread=TRUE)
dadaRs <- dada(derepRs, err=errR, multithread=TRUE)
# 检查,这里是检测正向的第一个样本
dadaFs[[1]]
## dada-class: object describing DADA2 denoising results
## 127 sequence variants were inferred from 1979 input unique sequences.
## Key parameters: OMEGA_A = 1e-40, BAND_SIZE = 16, USE_QUALS = TRUE
这里返回的信息意思是,通过上面消除误差后,发现独特的序列有1979条,而经过dada算法,其中的127条被判定为真实的物种序列。 ion torrent数据可加入参数HOMOPOLYMER_GAP_PENALTY=-1, BAND_SIZE=32。
六、合并双端数据
mergers <- mergePairs(dadaFs, derepFs, dadaRs, derepRs, verbose=TRUE)
# 栗子:查看第一个样本
head(mergers[[1]])
没有overlap的数据建议加入参数justConcatenate=TRUE。
七、构造列表
现在构建的表格是amplicon sequence variant(ASV)表格。可以认为是OTU表格的升级版。
seqtab <- makeSequenceTable(mergers)
dim(seqtab)
# 检测长度分布
table(nchar(getSequences(seqtab)))
dim出来的第二个值为扩增子序列个数。table(nchar())则统计出了每个读长下有多少个扩增子序列。
八、去除嵌合体
seqtab.nochim <- removeBimeraDenovo(seqtab, method="consensus", multithread=TRUE, verbose=TRUE)
dim(seqtab.nochim)
sum(seqtab.nochim)/sum(seqtab)
# 这个容易理解,在这些样本里,我得到的结果是剩下96.3%
如果,进行这一步之后,被去除了很多很多。那就说明原始数据有问题。大概的原因是原始数据中模糊核苷酸的引物序列还没被去除。
九、检查
getN <- function(x) sum(getUniques(x))
track <- cbind(
out, sapply(dadaFs, getN), sapply(dadaRs, getN), sapply(mergers, getN),
rowSums(seqtab.nochim)
)
# 如果是单样本,把每个sapply改为getN,栗子:sapply(dadaFs, getN)改为getN(dadaFs)
colnames(track) <- c("input", "filtered", "denoisedF", "denoisedR", "merged", "nonchim")
rownames(track) <- sample.names
head(track)
只要每一步的数据量没有一个大幅度的下降,就说明操作没问题。
十、物种分类
DADA2包提供了基于贝叶斯方法的分类。 assignTaxonomy函数将一组序列作为输入,再利用已知分类的参考序列的训练集,进行输出具有可信度的分类。 提供的训练集和参考序列包括了常用的GreenGenes、Silva、RDP等。这里使用Silva,因为比较新。 目前最新的版本v132可在这里下载。 要把训练集和参考序列都下了!
# 用训练集,将序列分类
taxa <- assignTaxonomy(seqtab.nochim, "silva_nr_v132_train_set.fa.gz", multithread=TRUE)
# 分好类之后,用参考序列,填上物种名字
taxa <- addSpecies(taxa, "silva_species_assignment_v132.fa.gz")
# 检查
taxa.print <- taxa
rownames(taxa.print) <- NULL
head(taxa.print)
如果结果类似这样:Eukaryota NA NA NA NA NA, 可能是因为序列与参考序列相反,修改下面这个参数:assignTaxonomy(…, tryRC=TRUE),可能可以解决问题。
十一、画各种图
还没搞懂!溜了溜了。 之后再来研究用phyloseq和ggplot2画图。