强推这篇文章,非常详尽,各种图例,眼花缭乱。 Analysis of single cell RNA-seq data
单细胞测序就是获取单个细胞遗传信息的测序技术,能够检出混杂样品测序所无法得到的异质性信息。
安装R包
SC3是一个非常好的用来分析单细胞测序的R包。
source("https://bioconductor.org/biocLite.R")
biocLite("SC3")
biocLite("scater")
biocLite("SingleCellExperiment")
该R包有些参数是依赖perl的,所以还需要有perl的环境。
前数据处理
单细胞测序,如果原始数据只有一个fastq,要先把每个细胞的测序结果分开。 首先使用trim_galore来去接头。 使用trim_galore之前,需要安装cutadapt。
pip install --user upgrade cutadapt
然后接下来是根据barcode来分开每个细胞的测序结果。 可以使用barcode_splitter
# 安装
pip install barcode_splitter
# 使用
barcode_splitter --bcfile barcodes.txt read1.fq.gz read2.fq.gz --idxread 2
当然,如果能确认一开始每个文件就是一个细胞的测序数据,就不用做这一步了。
这里推荐一个公开的不错的练手数据。GSE36552 一共是124个细胞,每个细胞单独为一个fq文件。
然后就是比对还有统计counts数/RPKM。 这里推荐参考上次写的RNA-seq流程,查看Hisat2用法,查看featuteCounts用法。
得到的结果,就是一个counts矩阵了。
使用SC3进行分析
这里是使用RPKM值的分析流程。
library(SingleCellExperiment)
library(SC3)
library(scater)
这里使用的是yan的数据集,作为SC3的示例数据。 这个数据是人胚胎发育不同时期的单细胞测序数据。 主要是两个表格 一个是样本信息
head(ann)
## cell_type1
## Oocyte..1.RPKM. zygote
## Oocyte..2.RPKM. zygote
## Oocyte..3.RPKM. zygote
## Zygote..1.RPKM. zygote
## Zygote..2.RPKM. zygote
## Zygote..3.RPKM. zygote
另外一个是RPKM值
yan[1:3, 1:3]
## Oocyte..1.RPKM. Oocyte..2.RPKM. Oocyte..3.RPKM.
## C9orf152 0.0 0.0 0.0
## RPS11 1219.9 1021.1 931.6
## ELMO2 7.0 12.2 9.3
实际操作时,根据需求选择究竟时用RPKM值还是用counts来做分析。 这里有文章说明了counts和FPKM值(pair ends数据为FPKM;single end数据为RPKM)的差别。 Counts vs. FPKMs in RNA-seq
# 创建SingleCellExperiment对象
sce <- SingleCellExperiment(
assays = list(
counts = as.matrix(yan),
logcounts = log2(as.matrix(yan) + 1)
),
colData = ann
)
# 创建基因名
rowData(sce)$feature_symbol <- rownames(sce)
# 去重复
sce <- sce[!duplicated(rowData(sce)$feature_symbol), ]
# 想要特别关注的基因
isSpike(sce, "ERCC") <- grepl("ERCC", rowData(sce)$feature_symbol)
# PCA
plotPCA(sce, colour_by = "cell_type1")
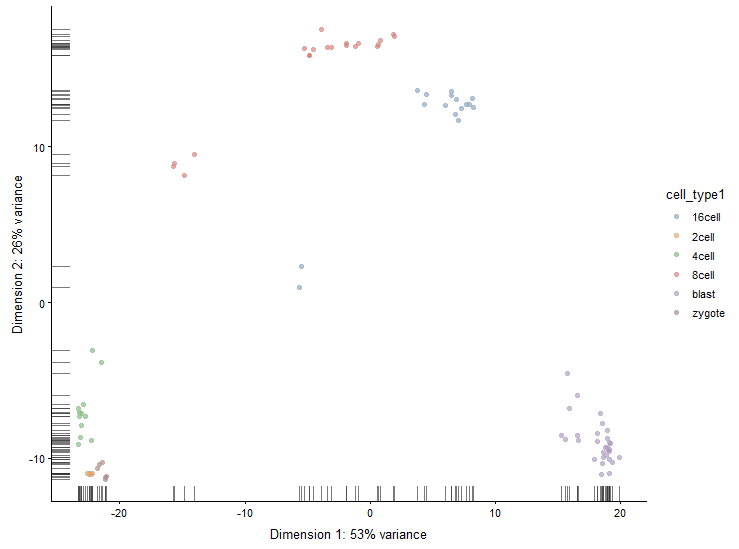
可以看出基本上胚胎发育不同时期的细胞已经有明显差异了。
运行SC3
将数据在ks(簇的数量)范围内探索数据的聚类,这里采取的范围是2:4。
sce <- sc3(sce, ks = 2:4, biology = TRUE)
输出结果到excel,这一步需要perl环境。
sc3_export_results_xls(sce)
和谐矩阵,计算聚类的和谐度(?),当对角线为红色而其他是蓝色时,说明聚类的效果好。
sc3_plot_consensus(
sce, k = 3,
show_pdata ="cell_type1"
)
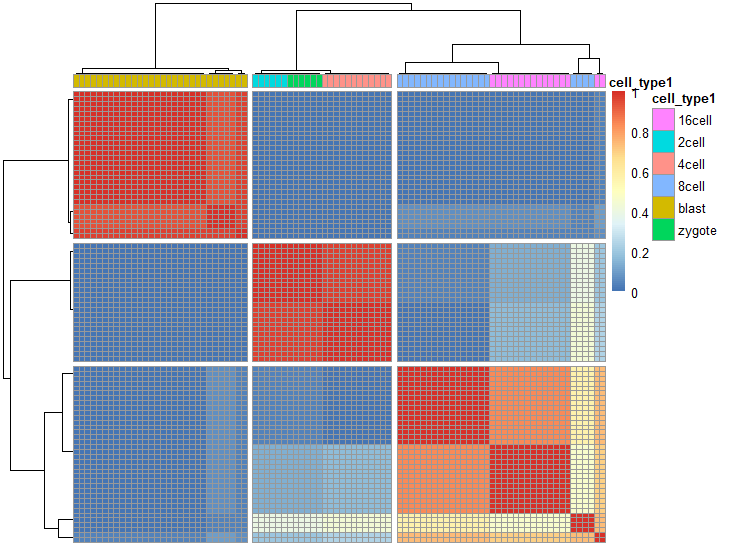
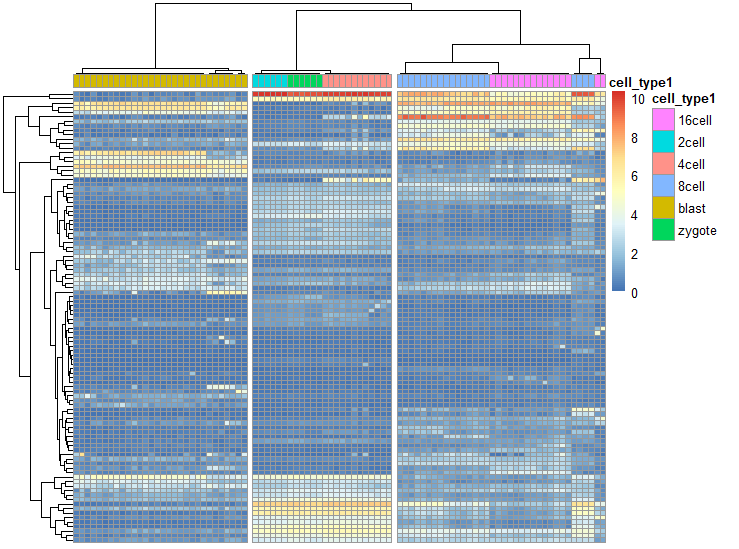
基因表达矩阵
sc3_plot_expression(
sce, k = 3,
show_pdata = "cell_type1"
)
差异表达
sc3_plot_de_genes(
sce, k = 3,
show_pdata = "cell_type1"
)
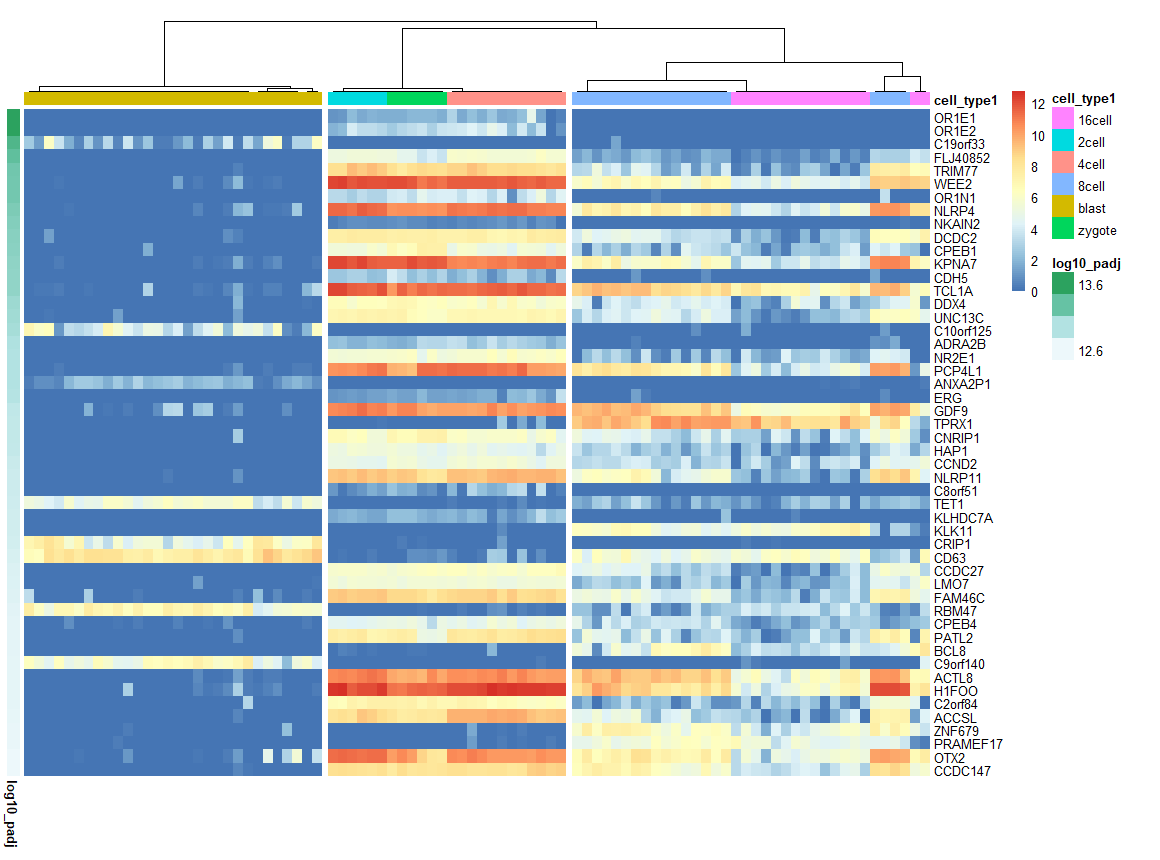
Marker基因
sc3_plot_markers(
sce, k = 3,
show_pdata = "cell_type1"
)
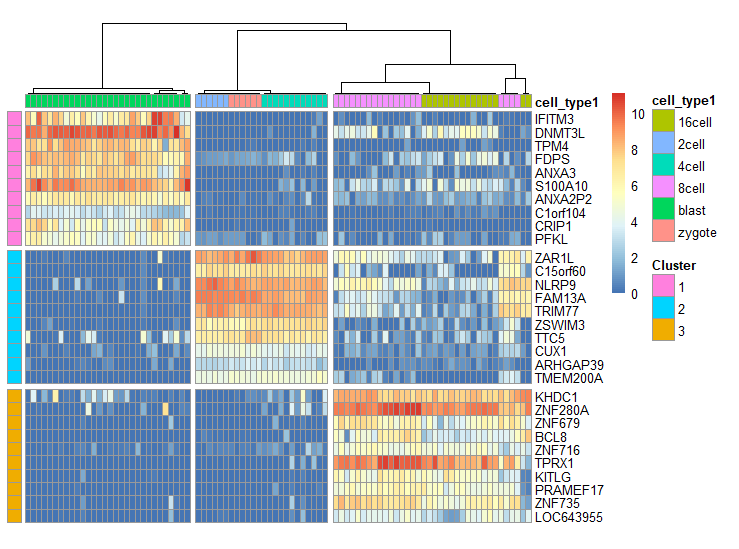
SC3这个包的特性很多,而且作者也在论文里讲了和另外一些包的分析结果对比。实际上,看作者前面的那篇终极教案,非常厉害。 争取一个月内学习完吧。
参考文献
Analysis of single cell RNA-seq data
SC3: consensus clustering of single-cell RNA-seq data
Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells