最近是深入的了解了一下BAM/SAM文件。
众所周知,SAM文件是比对后的文件,格式约定俗成,是一些大佬规定出来的格式,而BAM文件是SAM文件的二进制形式,为的是节省空间。
又众所周知,目前最好用的处理BAM文件和SAM文件的软件是samtools。
然后,对于python来说,有一个比较好用的包是pysam,根出同源。但是,pysam貌似和windows不兼容。
对于我这种需要跨平台工作的人来说,又找到了一个包,bamnostic。
SAM,其实是一个tab分割的文件。
SAM文件可以直接用
less test.sam
或者
cat test.sam
来查看。但是对于BAM文件这种二进制的格式,就需要用到samtools。 一般的
samtools view test.bam
当需要查看头信息时,就需要
samtools view -h test.bam
而同时samtools也支持了SAM和BAM文件的互转
samtools view -bSh test.sam > test.bam
samtools view -h test.bam > test.sam
接下来简单的说一下sam文件的格式
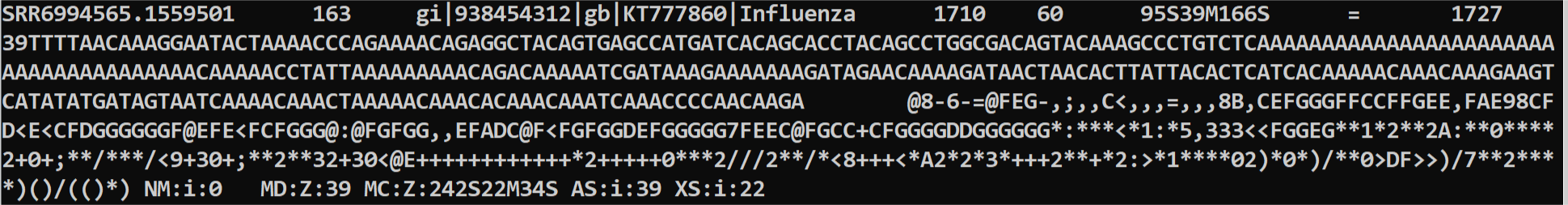
自己脑补tab分割。分别的意义是:
1 测序reads名称
2 flag值
3 比对到参考基因组的位置(染色体/contig)
4 从最左侧开始比对到的位置
5 MAPQ 比对的质量值 0-60表示从未比对上到最高,255表示结果不可用
6 CIGAR 表示比对状态,M表示匹配,I插入,D缺失,N可变剪切,S表示替换,H表示切除,例如这里就是这条reads前面95个碱基比对到其他地方,然后中间39个比对上了,后面166个又比对到其他地方了
7 reads二次比对结果,=表示没有二次结果;如果3和7都是*,表示没有结果;如果只有7是*,表示只比对到3的结果
8 position
9 文库插入片段长度
10 碱基序列
11 对应的碱基质量值
12一直到之后都是各种的tag
按照我之前做转基因分析来说,由于要找宿主与质粒合并的序列,所以我进行的其中一步操作是通过筛选SA这个tag来找嵌合的reads。就是通过下面这种简单粗暴的方式:
samtools view test.bam | grep "SA"
各种tag的解析可以点击这里找到。
再说bamnostic这个python包,其实文档写的挺详细。用起来我觉得算是够用,当然很多细节的功能不能完全满足,但是通过python的各种的type转换,还是能得到自己想要的结果的。
简单的安装
pip install bamnosic
使用
import bamnostic
bam = bs.AlignmentFile(bs.example_bam, 'rb')
这里有一些内置的函数可以快速的得到想要的结果,具体看文档! 但是,再不会,总会转成字符串,然后用最熟悉的做法呀:
for read in bam:
xxx = str(read).split("\t")